Materials Science
arXiv:cond-mat.mtrl-sci
Structural and mechanical properties of materials, synthesis, characterization methods.
Looking for a broader view? This category is part of:
Structural and mechanical properties of materials, synthesis, characterization methods.
Looking for a broader view? This category is part of:
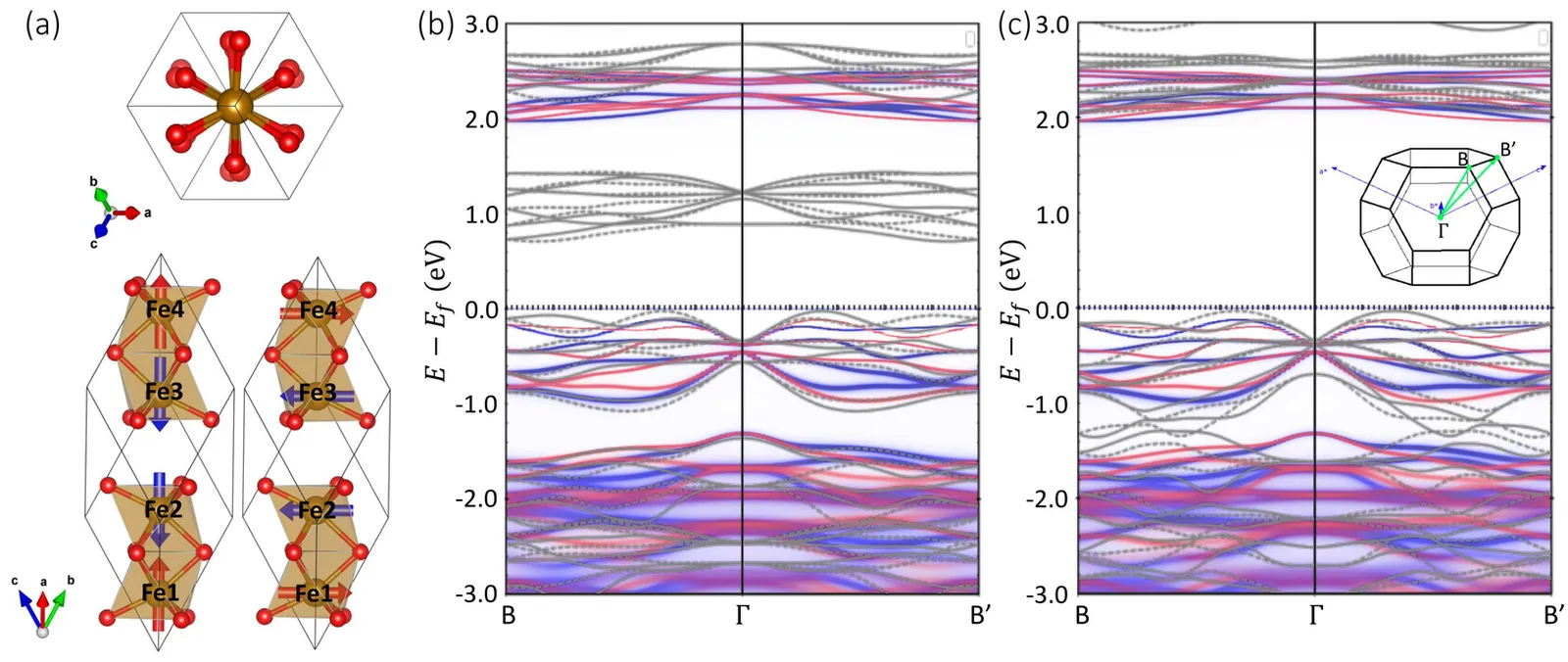
Hematite $α$-Fe$_2$O$_3$ is a $g$-wave altermagnetic material, which has an easy-axis phase and easy-plane weak ferromagnetic phase below and above Morin transition temperature, respectively. The presence of these phases renders it a good candidate to study the characteristic spin splitting in altermagnets under the impacts of relativistic effect and finite temperature. In this regard, we have calculated the band structure of $α$-Fe$_2$O$_3$ based on density functional theory (DFT) which also considers the Hubbard-U correction and spin-orbit coupling (SOC) effects. Additionally, the charge self-consistent DFT + dynamical mean-field theory (DMFT) calculations have been performed at finite temperatures. It is found that the altermagnetic spin splitting in $α$-Fe$_2$O$_3$ preserves with either SOC or temperature effect taken into account. Furthermore, we present a numerical simulation of the x-ray magnetic circular dichroism (XMCD) of the L$_{2,3}$ edge of Fe using a combination of DFT with multiplet ligand-field theory (MLFT). In terms of the different Néel vectors present in $α$-Fe$_2$O$_3$, we calculate the x-ray absorption spectroscopy (XAS) of the L$_{2,3}$ edge of Fe in the form of conductivity tensor and analyze the XMCD response from a perspective of symmetry. A characteristic XMCD line shape is expected when the Néel vector is along [010] direction (magnetic point group $2^\prime/m^\prime$) and the light propagation vector is perpendicular to the Néel vector, which can be further distinguished from the XMCD response originated from weak ferromagnetism with the light propagation vector parallel to the Néel vector.
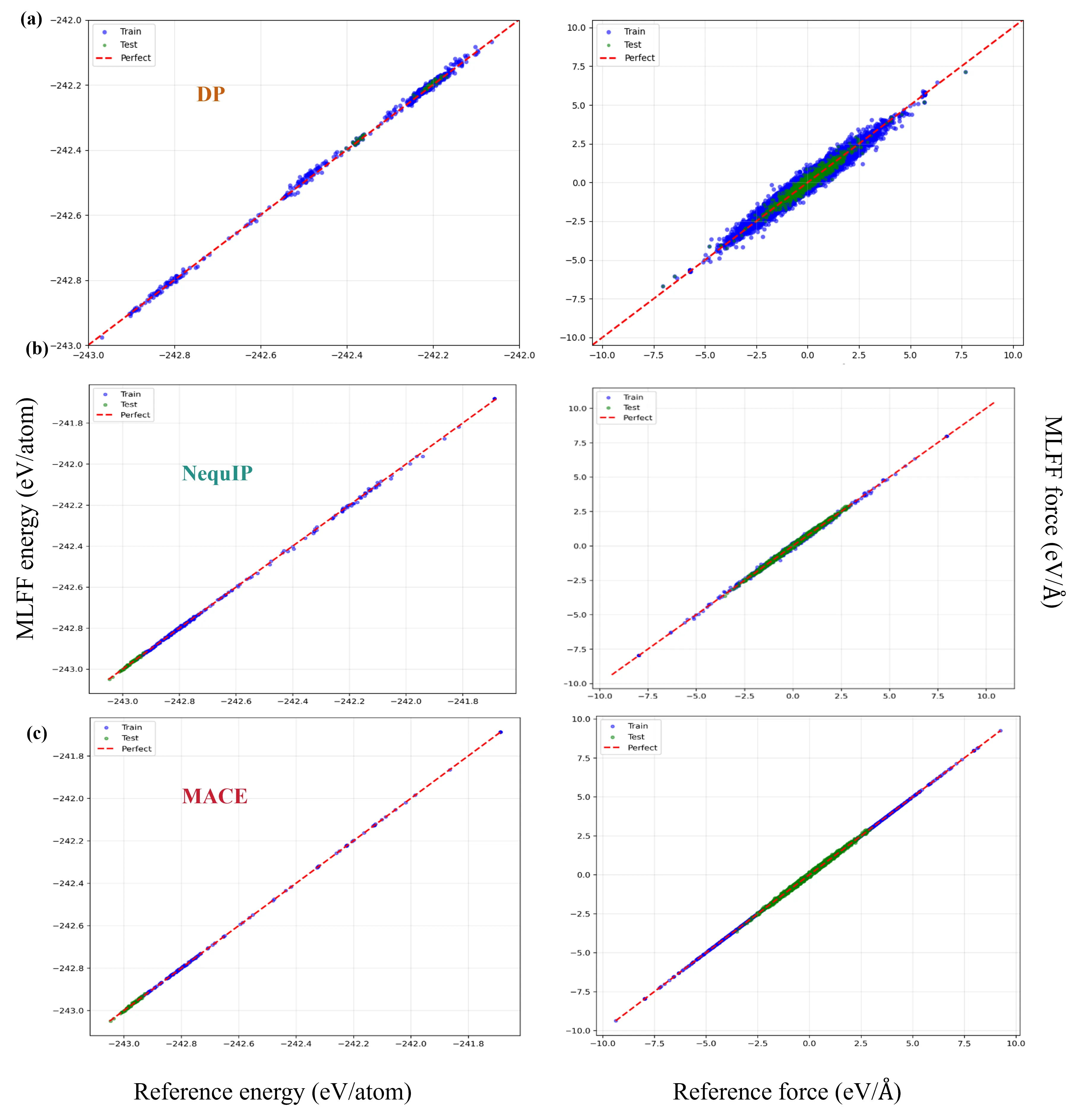
With the rapid advancement of machine learning techniques for materials simulations, machine-learned force fields (MLFFs) have become a powerful tool that complements first-principles calculations by enabling high-accuracy molecular dynamics simulations over extended timescales. Typically, MLFFs are trained on data generated from density functional theory (DFT) using a specific exchange-correlation (XC) functional, with the goal of reproducing DFT-level properties. However, the uncertainties in MLFF-based simulations--arising from variations in both MLFF model architectures and the choice of XC functionals--remain insufficiently understood. In this work, we construct MLFF models of different architectures trained on DFT data from both semilocal and hybrid functionals to describe Li$^+$ diffusion in the solid-state electrolyte Li$_6$PS$_5$Cl. We systematically investigate how different XC functionals influence the Li$^+$ diffusion coefficient. To reduce statistical uncertainty, the mean squared displacements are averaged over 300 independent molecular dynamics (MD) trajectories of 70 ps each, yielding statistical variations below $1\%$. This enables a clear assessment of the respective influences of the functional and the MLFF model. Due to its tendency to underestimate band gaps and migration barriers, the semilocal functional predicts consistently higher Li$^+$ diffusion coefficients, compared to the hybrid functional. Furthermore, comparisons among various neural network methods reveal that the differences in predicted diffusion coefficients arising from different network architectures are of the same order of magnitude as those caused by different functionals, indicating that the choice of the network model itself substantially influences the MLFF predictions. This observation calls from an urgent need for standardized protocols to minimize model-dependent biases in MLFF-based MD.
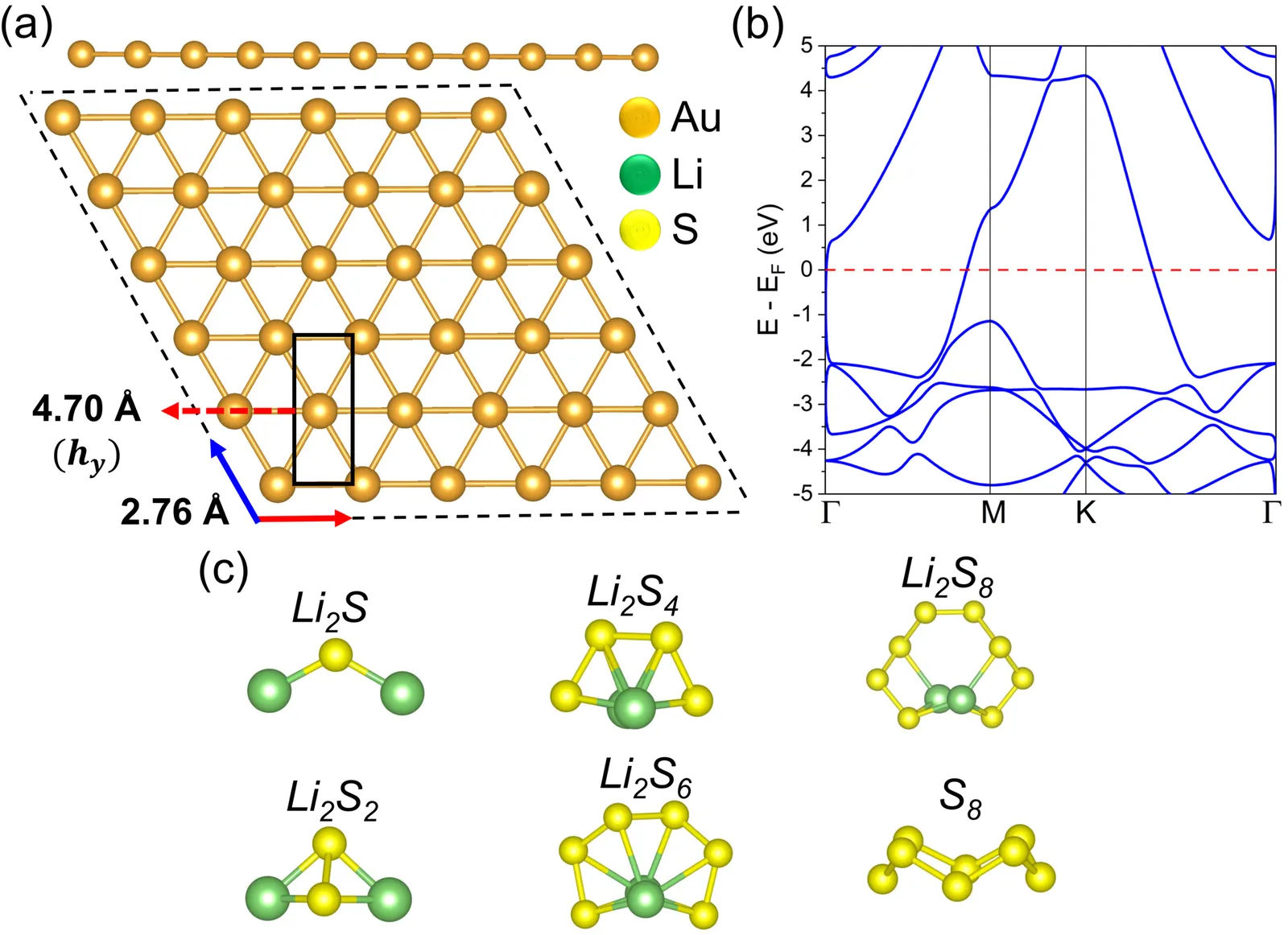
We use first-principles density functional theory to investigate how lithium sulfide and polysulfide clusters (Li2S, Li2S2, Li2S4, Li2S6, Li2S8, and S8) bind to Goldene, a new two-dimensional gold allotrope. All Li-S species exhibit robust binding to Goldene. The adsorption energies range from -4.29 to -1.90 eV. S8 that is alone interacts much less strongly. Charge density difference and Bader analyses indicate that substantial charge is transferred to the substrate, with a maximum 0.92 e for Li-rich clusters. This transfer induces polarization at the interface and shifts the work function to 5.30-5.52 eV. Projected density-of-states calculations indicate that Au-d and S-p states strongly mix near the Fermi level. This hybridization indicates that the electronic coupling is strong. Based on these results, the reaction free-energy profile for the stepwise conversion of S8 to Li2S on Goldene is thermodynamically favorable. The overall stabilization is -3.64 eV, and the rate-determining barrier for the Li2S2 -> Li2S step is 0.47 eV. This shows that Goldene is an effective surface for anchoring and mediating lithium polysulfide reactions.
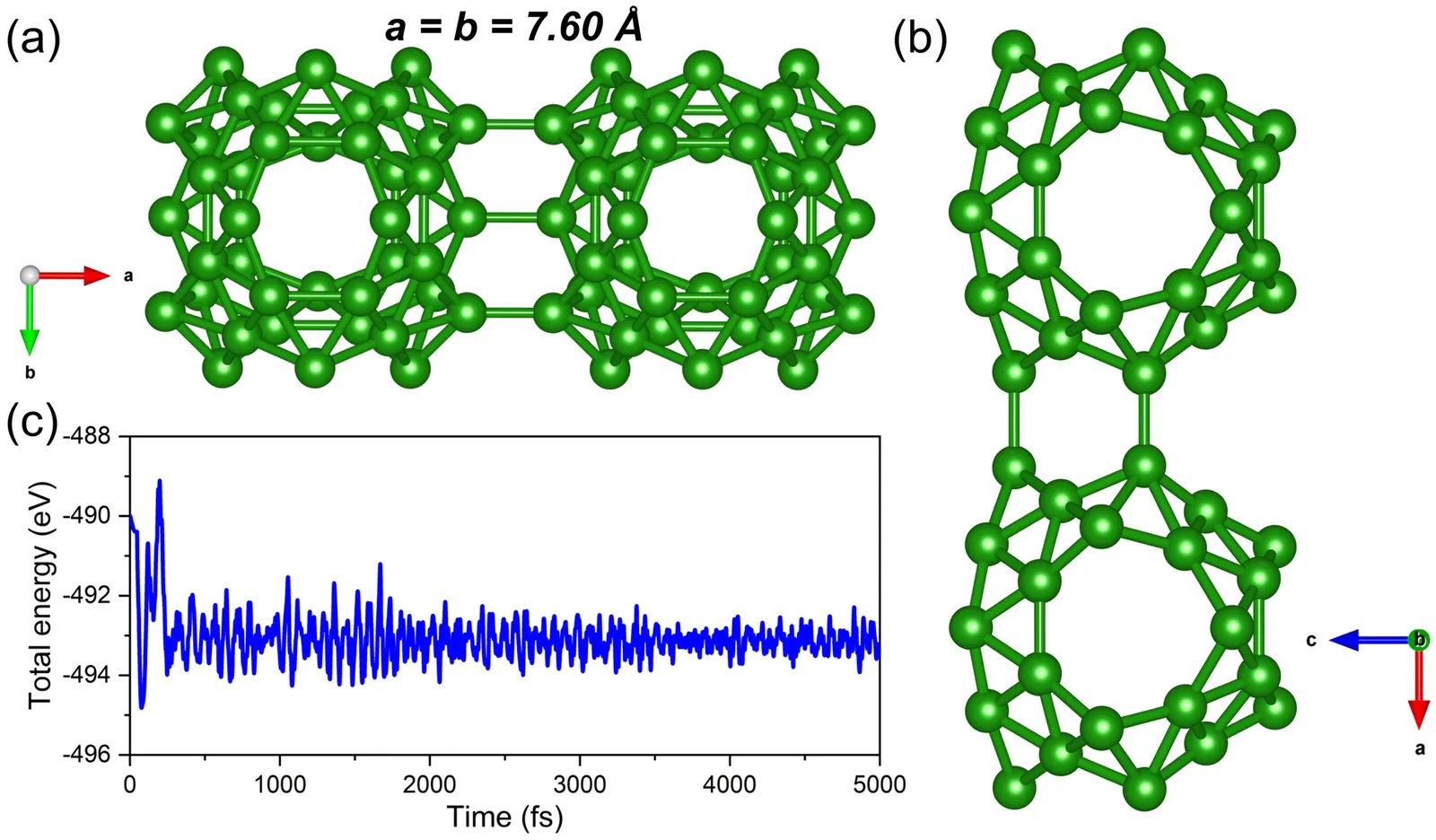
Two-dimensional (2D) boron-based materials have gained increasing interest due to their exceptional physicochemical properties and potential technological applications. In this way, borospherenes, a 2D Boron-based fullerene-like lattice (2D-B40), are explored due to their potential for capturing and detecting toxic gases, such as CO, NO, NH3, and SO2. Therefore, density functional theory simulations were carried out to explore the adsorption energy and the distinct interaction regimes, where CO exhibits weak physisorption (-0.16 eV), while NO (-2.24 eV), NH3 (-1.47 eV), and SO2 (-1.51 eV) undergo strong chemisorption. Bader charge analysis reveals significant electron donation from 2D-B40 to NO and electron acceptance from SO2. These interactions cause measurable shifts in work function, with SO2 producing the most significant modulation (14.6%). Remarkably, ab initio molecular dynamics simulations (AIMD) reveal spontaneous SO2 decomposition at room temperature, indicating dual functionality for both sensing and environmental remediation. Compared to other boron-based materials, such as chi3-borophene, beta12-borophene, and B40 fullerene, 2D-B40 exhibits superior gas affinity, positioning it as a versatile platform for the detection and capture of toxic gases.
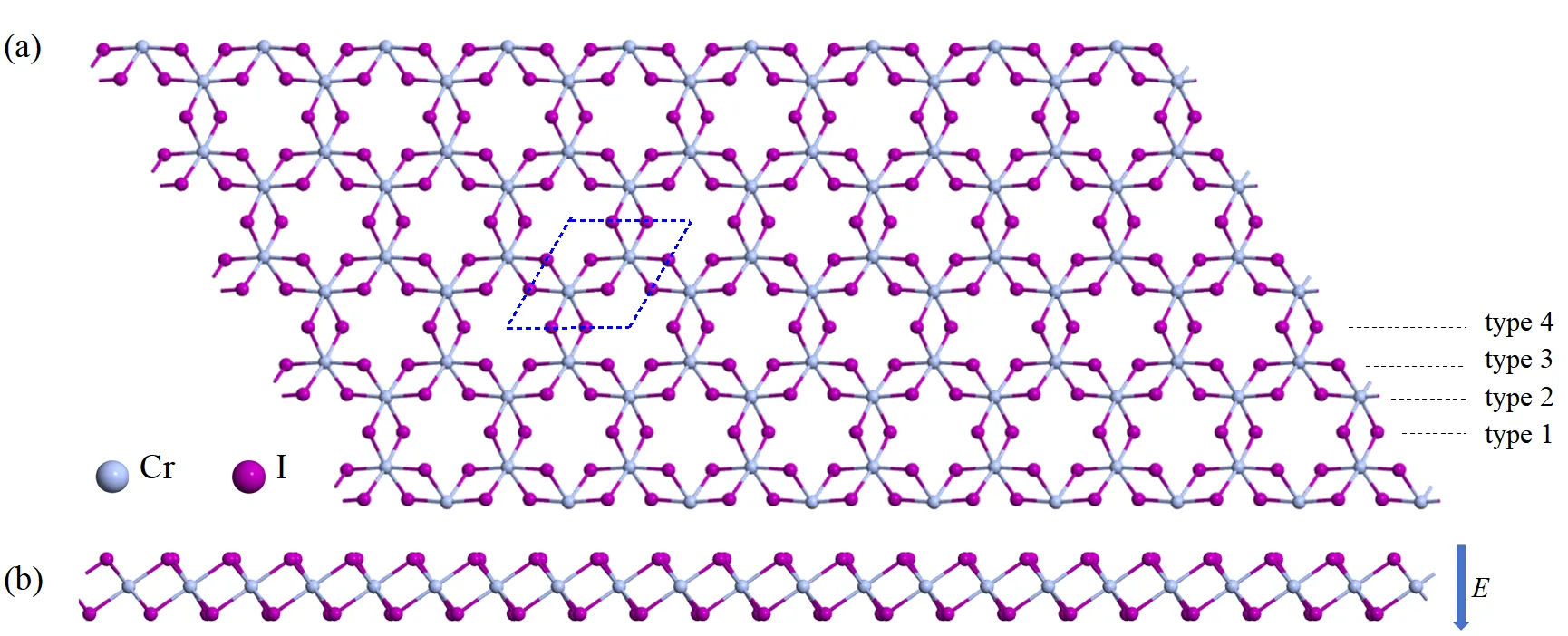
The successful synthesis of few-layer CrI3 has opened new avenues for research in two-dimensional magnetic materials. Owing to its simple crystal structure and excellent physical properties, layered CrI3 has been extensively studied in magneto-optical effects, excitons, tunneling transport, and novel memory devices. However, the most current theoretical studies rely heavily on the first-principles calculations, and a general analytical theoretical framework, particularly for electric-field modulation and transport properties, is still lacking. In this work, using a 28-band tight-binding model combined with linear response theory, we systematically investigate the optoelectronic response for monolayer CrI3 and its nanoribbons. The results demonstrate that: (1) a vertical electric field can selectively close the band gap of one spin channel while the other remains insulating, resulting a transition to an half-metallic state; (2) the electric field dynamically shifts the optical transition peaks, providing a theoretical basis for extracting band parameters from experimental photoconductivity spectra; (3) nanoribbons with different edge morphologies exhibit distinct edge-state distributions and electronic properties, indicating that optical transition can be dynamically modualted through edge design. The theoretical model developed in this study, which can describe external electric field effect, offers an efficient and flexible approach for analytically investigating the CrI3 family and related materials. This model overcomes the limitations of first-principles methods and provides a solid foundation for designing spintronic and optoelectronic devices controlled by electric fields and edge effect.
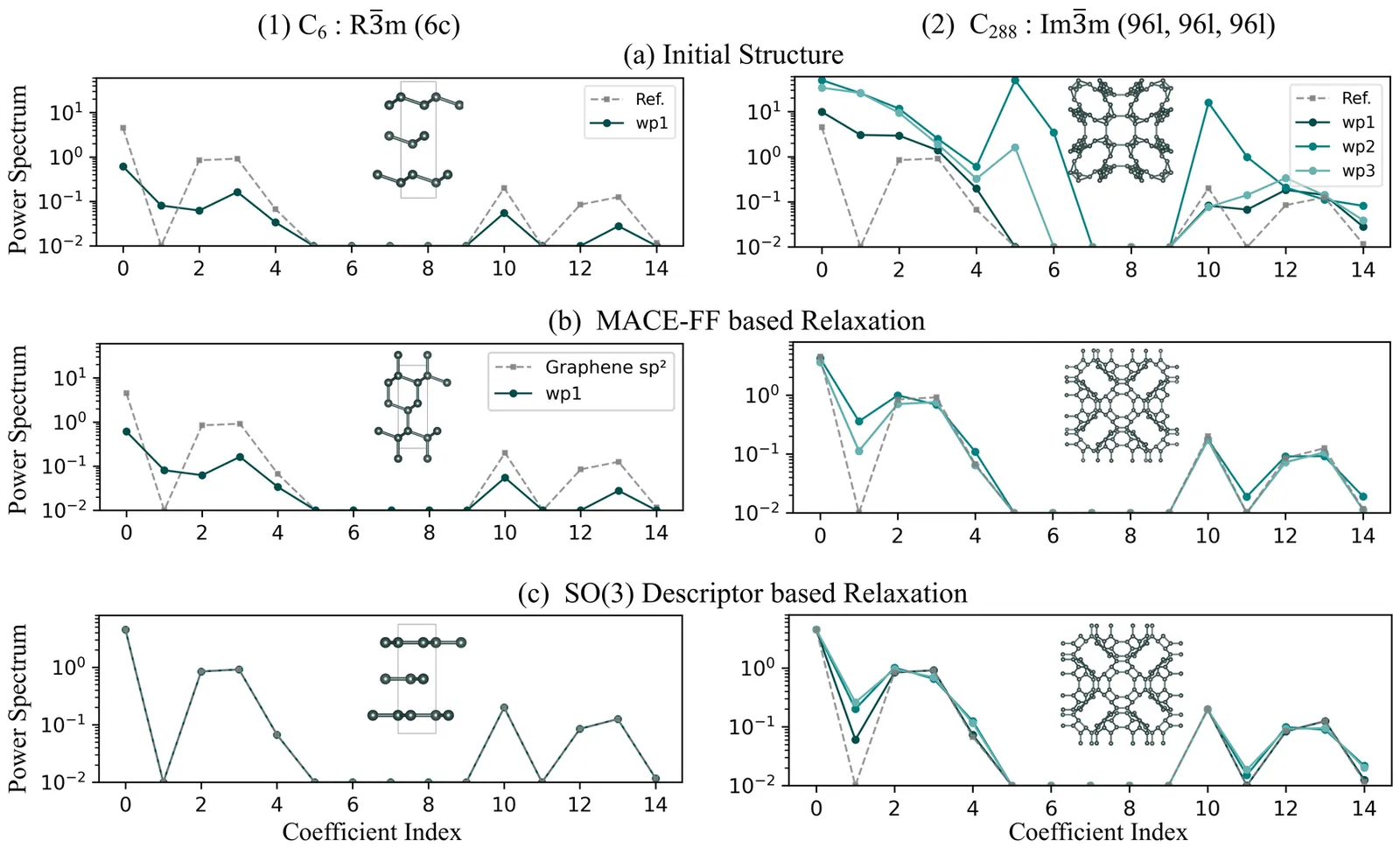
We present a materials generation framework that couples a symmetry-conditioned variational autoencoder (CVAE) with a differentiable SO(3) power spectrum objective to steer candidates toward a specified local environment under the crystallographic constraints. In particular, we implement a fully differentiable pipeline that performs batch-wise optimization on both direct and latent crystallographic representations. Using the GPU acceleration, the implementation achieves about fivefold speed compared to our previous CPU workflow, while yielding comparable outcomes. In addition, we introduce the optimization strategy that alternatively performs optimization on the direct and latent crystal representations. This dual-level relaxation approach can effectively overcome local barrier defined by different objective gradients, thus increasing the success rate of generating complex structures satisfying the targe local environments. This framework can be extended to systems consisting of multi-components and multi-environments, providing a scalable route to generate material structures with the target local environment.
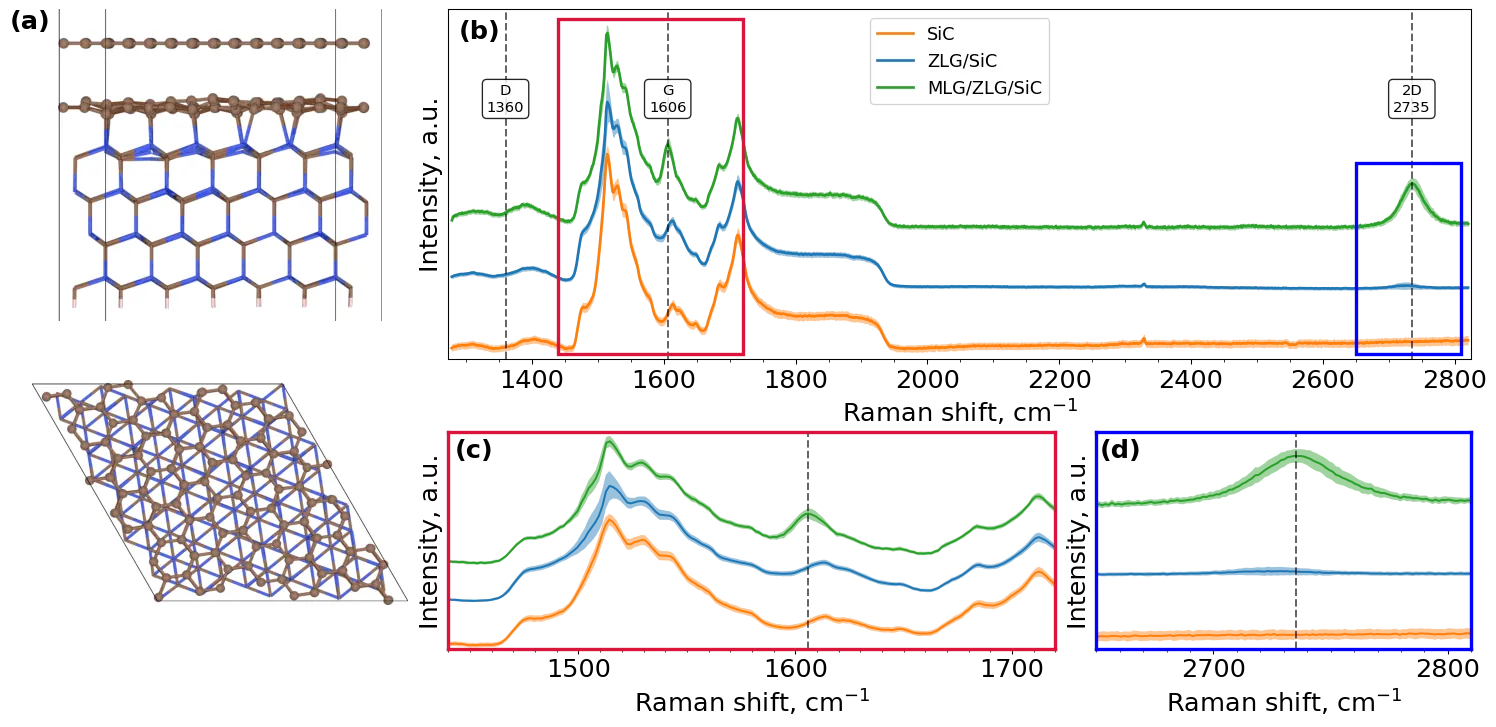
Raman spectroscopy is a key tool for graphene characterization, yet its application to graphene grown on silicon carbide (SiC) is strongly limited by the intense and variable second-order Raman response of the substrate. This limitation is critical for buffer layer graphene, a semiconducting interfacial phase, whose vibrational signatures are overlapped with the SiC background and challenging to be reliably accessed using conventional reference-based subtraction, due to strong spatial and experimental variability of the substrate signal. Here we present SpectraFormer, a transformer-based deep learning model that reconstructs the SiC Raman substrate contribution directly from post-growth partially masked spectroscopic data without relying on explicit reference measurements. By learning global correlations across the entire Raman shift range, the model captures the statistical structure of the SiC background and enables accurate reconstruction of its contribution in mixed spectra. Subtraction of the reconstructed substrate signal reveals weak vibrational features associated with ZLG that are inaccessible through conventional analysis methods. The extracted spectra are validated by ab initio vibrational calculations, allowing assignment of the resolved features to specific modes and confirming their physical consistency. By leveraging a state-of-the-art attention-based deep learning architecture, this approach establishes a robust, reference-free framework for Raman analysis of graphene on SiC and provides a foundation, compatible with real-time data acquisition, to its integration into automated, closed-loop AI-assisted growth optimization.
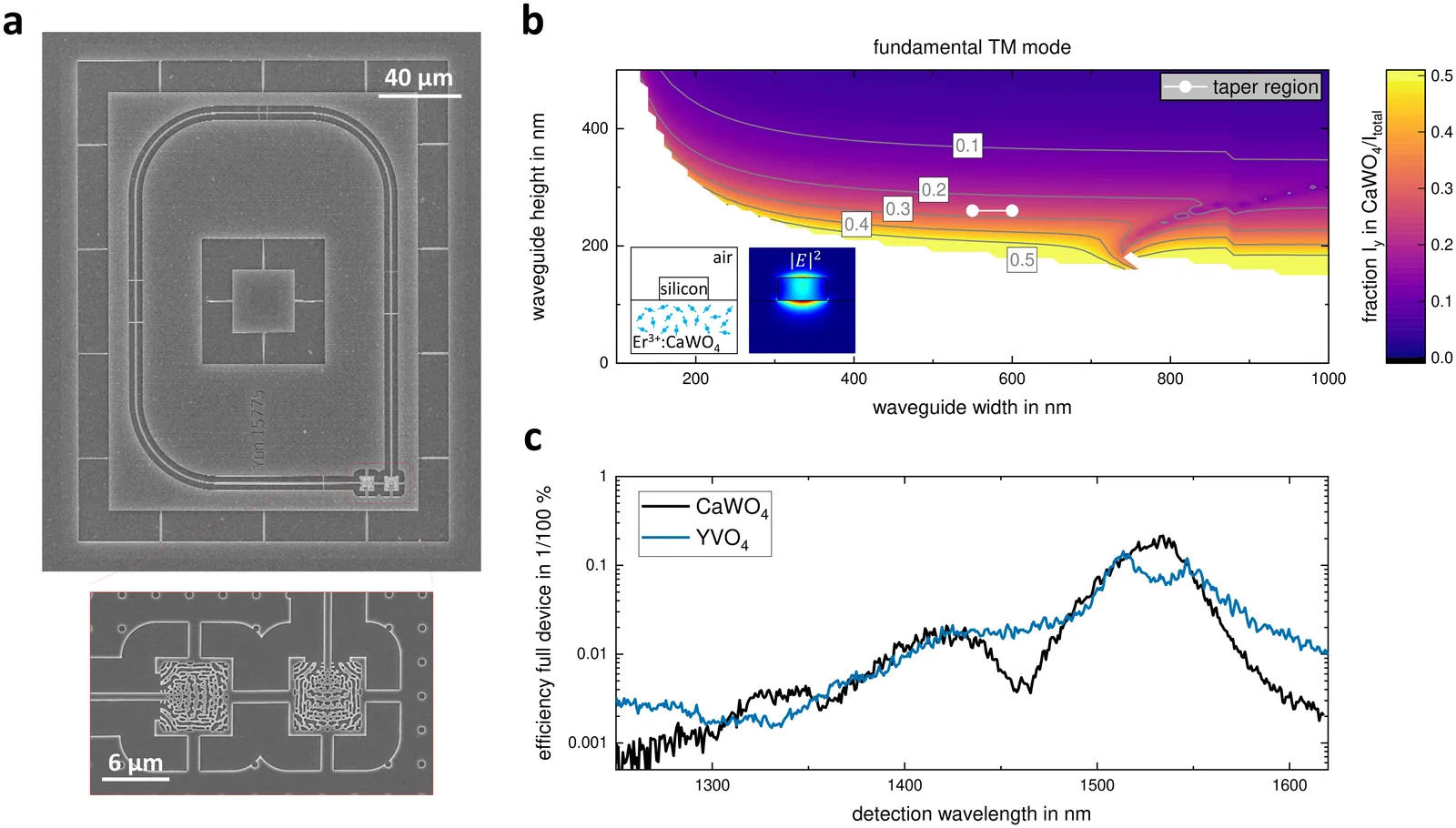
We present an optical study of near-surface Er$^{3+}$ ensembles in waveguide-integrated CaWO$_4$ and YVO$_4$, investigating how nanophotonic coupling modifies rare-earth spectroscopy. In particular, we compare bulk excitation with evanescently coupled TE and TM waveguide modes. In Er$^{3+}$:CaWO$_4$, we observe a pronounced polarization-dependent surface effect. TE-coupled spectra closely reproduce bulk behavior. In contrast, TM coupling induces strong inhomogeneous broadening and an asymmetric low-energy shoulder of the site S1 Y1Z1 transition, with linewidths exceeding those of the bulk by more than a factor of four. Temperature-dependent measurements and surface termination studies indicate that surface charges are the dominant mechanism. Er$^{3+}$:YVO$_4$ remains largely unaffected by mode polarization, and surface termination leads only to minor spectral shifts. These observations suggest that non-charge-neutral rare-earth systems are more susceptible to surface-induced decoherence sources than charge-neutral hosts.
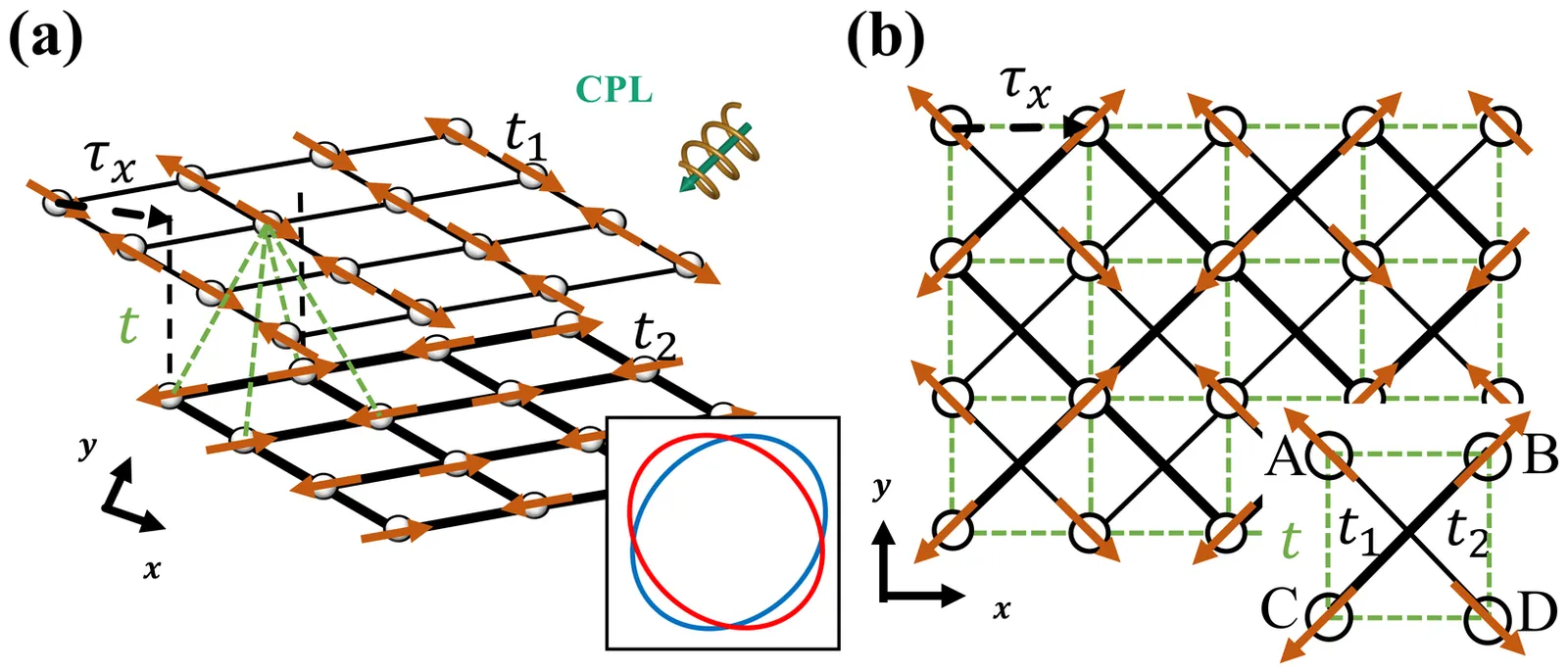
When a coplanar antiferromagnet (AFM) with $xy$-plane magnetic moments exhibits a spin-split band structure and unidirectional spin polarization along $z$, the spin polarization is forced to be an odd function of momentum by the fundamental symmetry $[\bar{C}_{2z}\|\mathcal{T}]$. Coplanar AFMs displaying such odd-parity unidirectional spin splittings are known as odd-parity magnets. In this work, we propose the realization of their missing even-parity counterparts. We begin by deriving the symmetry conditions required for an even-parity, out-of-plane spin splitting. We then show that irradiating a spin-degenerate coplanar AFM with circularly polarized light lifts the $[\bar{C}_{2z}|\mathcal{T}]$ constraint, dynamically generating this even-parity state. Specifically, the light-induced unidirectional spin splitting exhibits a $d$-wave texture in momentum space, akin to that of a $d$-wave altermagnet. We prove this texture's robustness against spin canting and show it yields a unique clover-like angular dependence in the Drude spin conductivity. Our work demonstrates that optical driving can generate novel spin-split phases in coplanar AFMs, thereby diversifying the landscape of materials exhibiting distinct spin splittings.
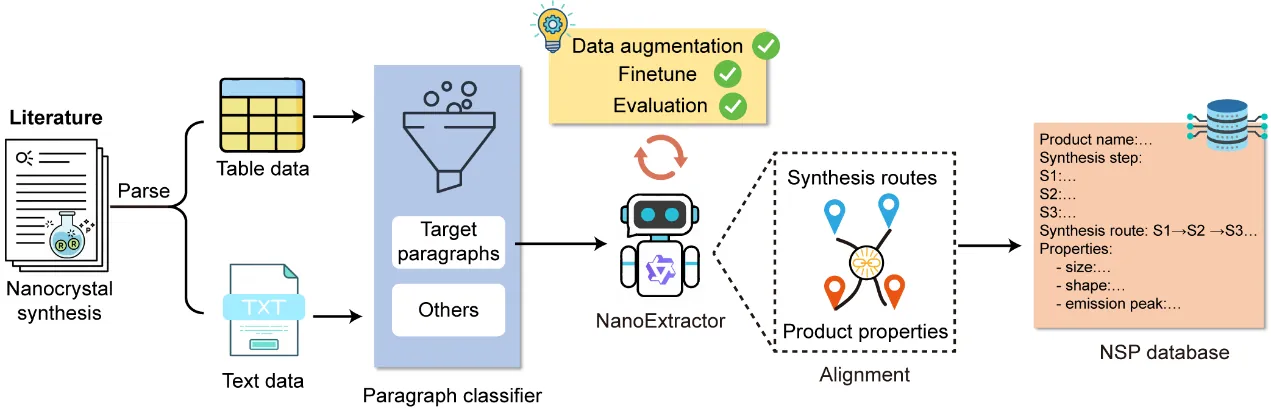
The synthesis of nanocrystals has been highly dependent on trial-and-error, due to the complex correlation between synthesis parameters and physicochemical properties. Although deep learning offers a potential methodology to achieve generative inverse design, it is still hindered by the scarcity of high-quality datasets that align nanocrystal synthesis routes with their properties. Here, we present the construction of a large-scale, aligned Nanocrystal Synthesis-Property (NSP) database and demonstrate its capability for generative inverse design. To extract structured synthesis routes and their corresponding product properties from literature, we develop NanoExtractor, a large language model (LLM) enhanced by well-designed augmentation strategies. NanoExtractor is validated against human experts, achieving a weighted average score of 88% on the test set, significantly outperforming chemistry-specialized (3%) and general-purpose LLMs (38%). The resulting NSP database contains nearly 160,000 aligned entries and serves as training data for our NanoDesigner, an LLM for inverse synthesis design. The generative capability of NanoDesigner is validated through the successful design of viable synthesis routes for both well-established PbSe nanocrystals and rarely reported MgF2 nanocrystals. Notably, the model recommends a counter-intuitive, non-stoichiometric precursor ratio (1:1) for MgF2 nanocrystals, which is experimentally confirmed as critical for suppressing byproducts. Our work bridges the gap between unstructured literature and data-driven synthesis, and also establishes a powerful human-AI collaborative paradigm for accelerating nanocrystal discovery.
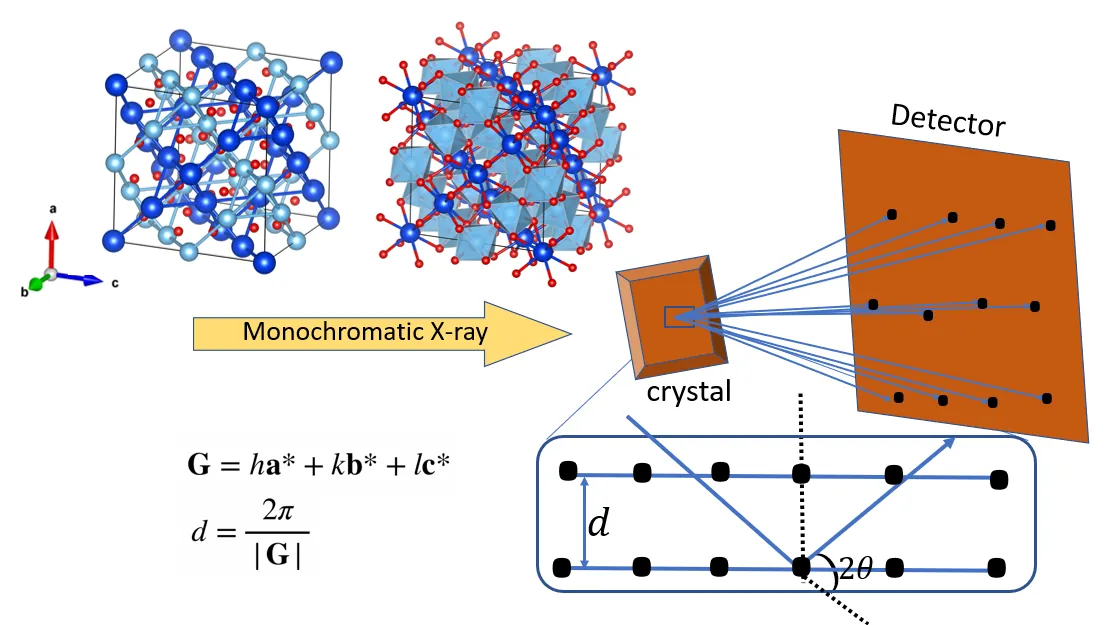
Physical properties and functionalities of materials are dictated by global crystal structures as well as local defects. To establish a structure-property relationship, not only the crystallographic symmetry but also quantitative knowledge about defects are required. Here we present a hybrid Machine Learning framework that integrates a physically-constrained variational-autoencoder (pcVAE) with different Bayesian Optimization (BO) methods to systematically accelerate and improve crystal structure refinement with resolution of defects. We chose the pyrochlore structured Ho2Ti2O7 as a model system and employed the GSAS2 package for benchmarking crystallographic parameters from Rietveld refinement. However, the function space of these material systems is highly nonlinear, which limits optimizers like traditional Rietveld refinement, into trapping at local minima. Also, these naive methods don't provide an extensive learning about the overall function space, which is essential for large space, large time consuming explorations to identify various potential regions of interest. Thus, we present the approach of exploring the high Dimensional structure parameters of defect sensitive systems via pretrained pcVAE assisted BO and Sparse Axis Aligned BO. The pcVAE projects high-Dimensional diffraction data consisting of thousands of independently measured diffraction orders into a lowD latent space while enforcing scaling invariance and physical relevance. Then via BO methods, we aim to minimize the L2 norm based chisq errors in the real and latent spaces separately between experimental and simulated diffraction patterns, thereby steering the refinement towards potential optimum crystal structure parameters. We investigated and compared the results among different pcVAE assisted BO, non pcVAE assisted BO, and Rietveld refinement.
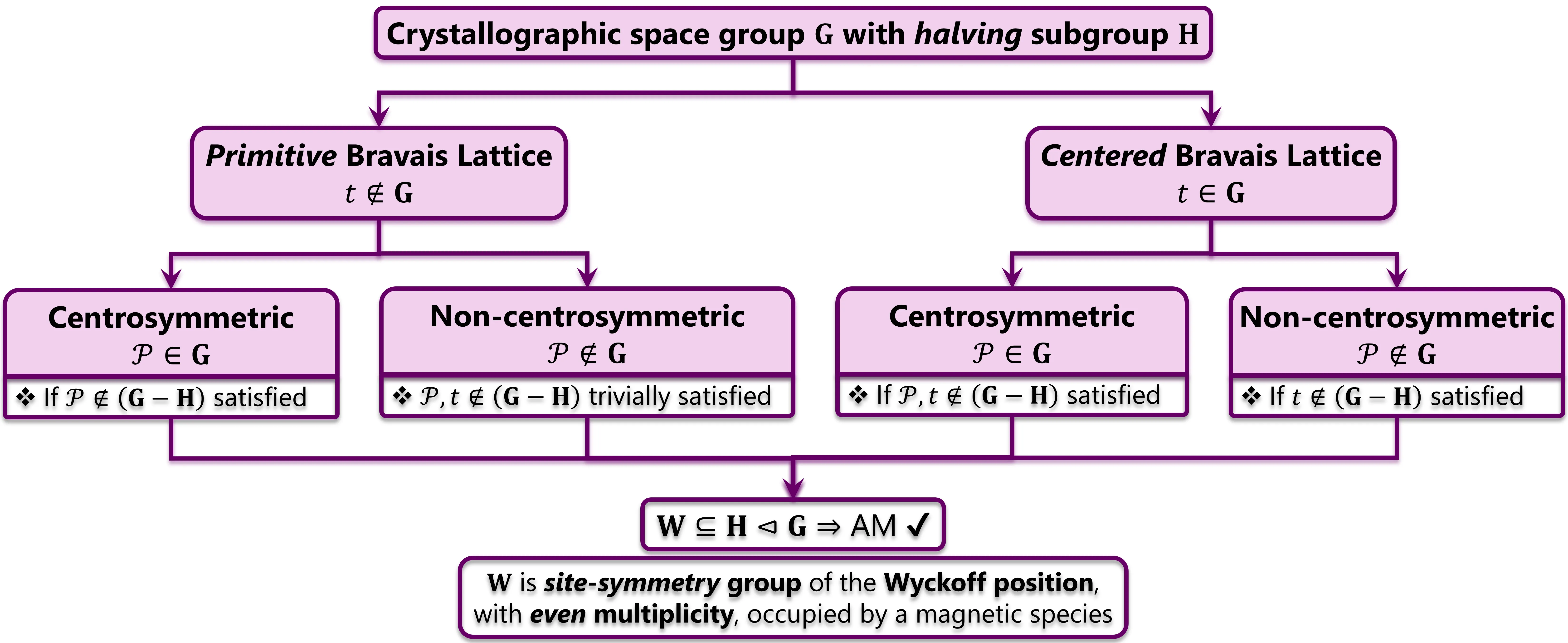
Recent years have seen a proliferation in investigations on Altermagnetism due to its exciting prospects both from an applications perspective and theoretical standpoint. Traditionally, altermagnets are distinguished from collinear antiferromagnets using the central concept of halving subgroups within the spin space group formalism. In this work, we propose the Fundamental Lemma of Altermagnetism (FLAM) deriving the exact conditions required for the existence of altermagnetic phase in a magnetic material on the basis of site-symmetry groups and halving subgroups for a given crystallographic space group. The spin group formalism further clubs ferrimagnetism with ferromagnetism since the same-spin and opposite-spin sublattices lose their meaning in the presence of multiple magnetic species. As a consequence of FLAM, we further propose a class of fully compensated ferrimagnets, termed as Alterferrimagnets (AFiMs), which can show alternating momentum-dependent spin-polarized non-relativistic electronic bands within the first Brillouin zone. We show that alterferrimagnetism is a generalization of traditional collinear altermagnetism where multiple magnetic species are allowed to coexist forming fully compensated magnetic-sublattices, each with individual up-spin and down-spin sublattices.
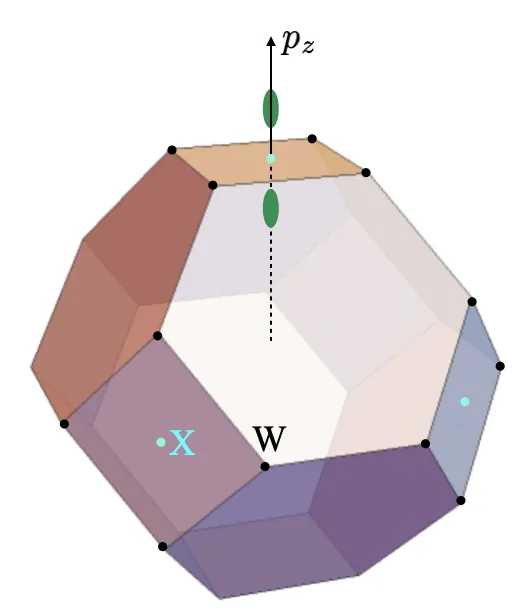
Increasing the valley splitting in Si-based heterostructures is critical for improving the performance of semiconductor qubits. This paper demonstrates that the two low-energy conduction band valleys are not independent parabolic bands. Instead, they originate from the X-point of the Brillouin zone, where they are interconnected by a degeneracy protected by the non-symmorphic symmetry of the diamond lattice. This semi-Dirac-node degeneracy gives rise to the $Δ_1$ and $Δ_{2'}$ bands, which constitute the valley degrees of freedom. By explicitly computing the two-component Bloch functions $X_1^\pm$, using the wave vector group at the X-point, we determine the transformation properties of the object $(X_1^+,X_1^-)$. We demonstrate that these properties are fundamentally different from those of a spinor. Consequently, we introduce the term "valleyor" to emphasize this fundamental distinction. The transformation properties of valleyors induce corresponding transformations of the Pauli matrices $τ_1,τ_2$ and $τ_3$ in the valley space. Determining these transformations allows us to classify possible external perturbations that couple to each valley Pauli matrix, thereby identifying candidates for valley-magnetic fields, ${\mathsf B}$. These fields are defined by a Zeeman-like coupling ${\mathsf B}\cdot\vecτ$ to the valley degree of freedom. In this way, we identify scenarios where an applied magnetic field $\vec B$ can leverage other background fields, such as strain, to generate a valley-magnetic field ${\mathsf B}$. This analysis suggests that beyond the well-known mechanism of potential scattering from Ge impurities, there exist additional channels (mediated by combinations of magnetic and strain-induced vector potentials) to control the valley degree of freedom
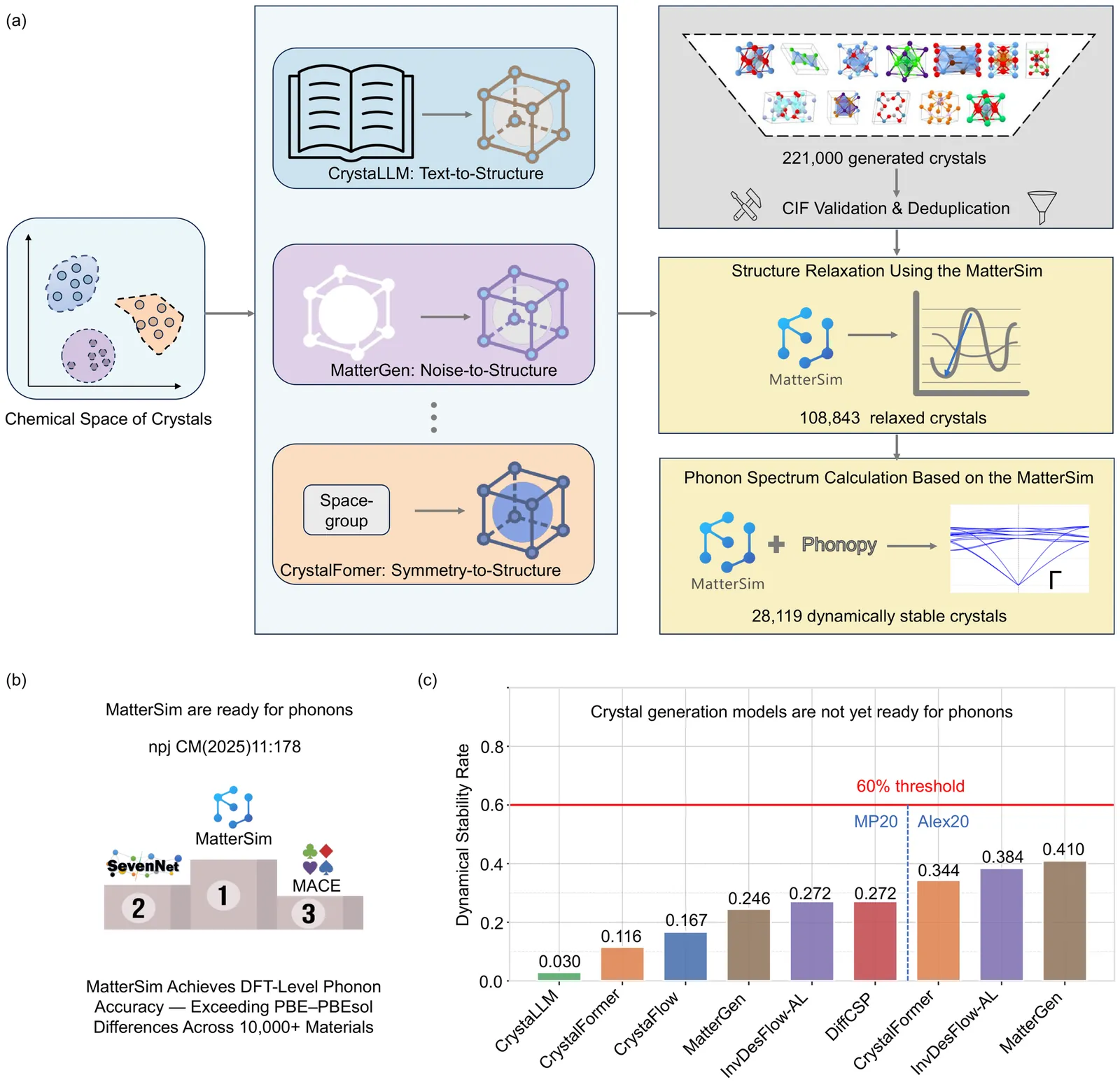
In this work, we introduce PhononBench, the first large-scale benchmark for dynamical stability in AI-generated crystals. Leveraging the recently developed MatterSim interatomic potential, which achieves DFT-level accuracy in phonon predictions across more than 10,000 materials, PhononBench enables efficient large-scale phonon calculations and dynamical-stability analysis for 108,843 crystal structures generated by six leading crystal generation models. PhononBench reveals a widespread limitation of current generative models in ensuring dynamical stability: the average dynamical-stability rate across all generated structures is only 25.83%, with the top-performing model, MatterGen, reaching just 41.0%. Further case studies show that in property-targeted generation-illustrated here by band-gap conditioning with MatterGen--the dynamical-stability rate remains as low as 23.5% even at the optimal band-gap condition of 0.5 eV. In space-group-controlled generation, higher-symmetry crystals exhibit better stability (e.g., cubic systems achieve rates up to 49.2%), yet the average stability across all controlled generations is still only 34.4%. An important additional outcome of this study is the identification of 28,119 crystal structures that are phonon-stable across the entire Brillouin zone, providing a substantial pool of reliable candidates for future materials exploration. By establishing the first large-scale dynamical-stability benchmark, this work systematically highlights the current limitations of crystal generation models and offers essential evaluation criteria and guidance for their future development toward the design and discovery of physically viable materials. All model-generated crystal structures, phonon calculation results, and the high-throughput evaluation workflows developed in PhononBench will be openly released at https://github.com/xqh19970407/PhononBench

Defects in atomically thin van der Waals materials have recently been investigated as sources of spin-photon entanglement with sensitivity to strain tuning. Unlike many two-dimensional materials, quasi-one-dimensional materials such as transition metal trichalcogenides exhibit in-plane anisotropy resulting in axis-dependent responses to compressive and tensile strains. Herein, we characterize the tunable spin and optical properties of intrinsic vacancy defects in titanium trisulfide (TiS3) and niobium trisulfide (NbS3) nanowires. Within our ab initio approach, we show that sulfur vacancies and divacancies (VS and VD , respectively) in TiS3 and NbS3 adopt strain-dependent defect geometries between in-plane strains of -3 % and 3 %. The calculated electronic structures indicate that both VS and VD possess in-gap defect states with optically bright electronic transitions whose position relative to the conduction and valence bands varies with in-plane strain. Further, our calculations predict that VS in TiS3 and VD in NbS3 exhibit transitions in their ground state spins; specifically, a compressive strain of 0.4 % along the direction of nanowire growth causes a shift from a triplet state to a singlet state for the VS defect in TiS3, whereas a tensile strain of 2.9 % along the same direction in NbS3 induces a triplet ground state with a zero-phonon line of 0.83 eV in the VD defect. Our work shows that the anisotropic geometry of TiS3 and NbS3 nanowires offers exceptional tunability of optically active spin defects that can be used in quantum applications.
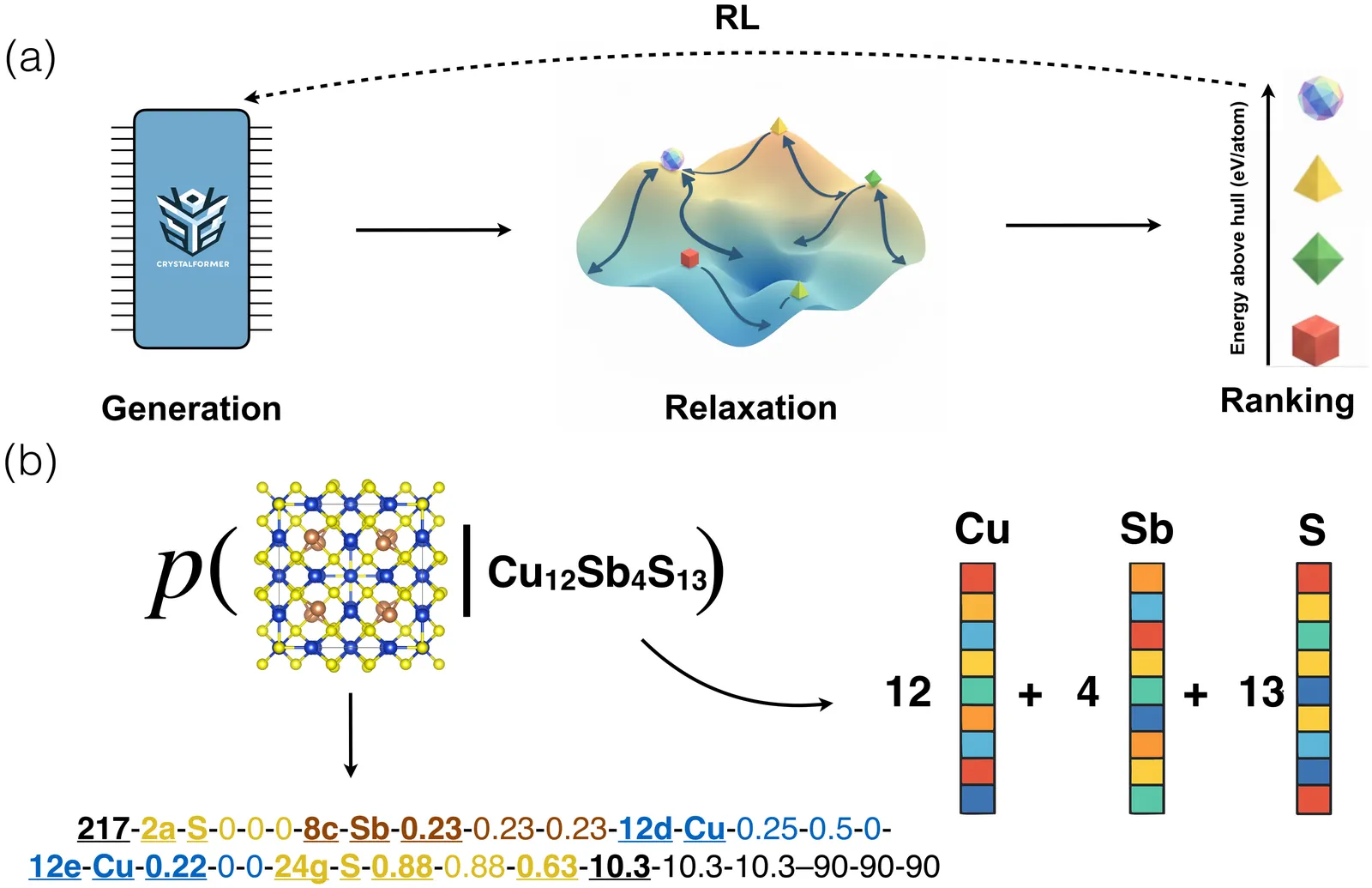
Crystal structure prediction is a fundamental problem in materials science. We present CrystalFormer-CSP, an efficient framework that unifies data-driven heuristic and physics-driven optimization approaches to predict stable crystal structures for given chemical compositions. The approach combines pretrained generative models for space-group-informed structure generation and a universal machine learning force field for energy minimization. Reinforcement fine-tuning can be employed to further boost the accuracy of the framework. We demonstrate the effectiveness of CrystalFormer-CSP on benchmark problems and showcase its usage via web interface and language model integration.
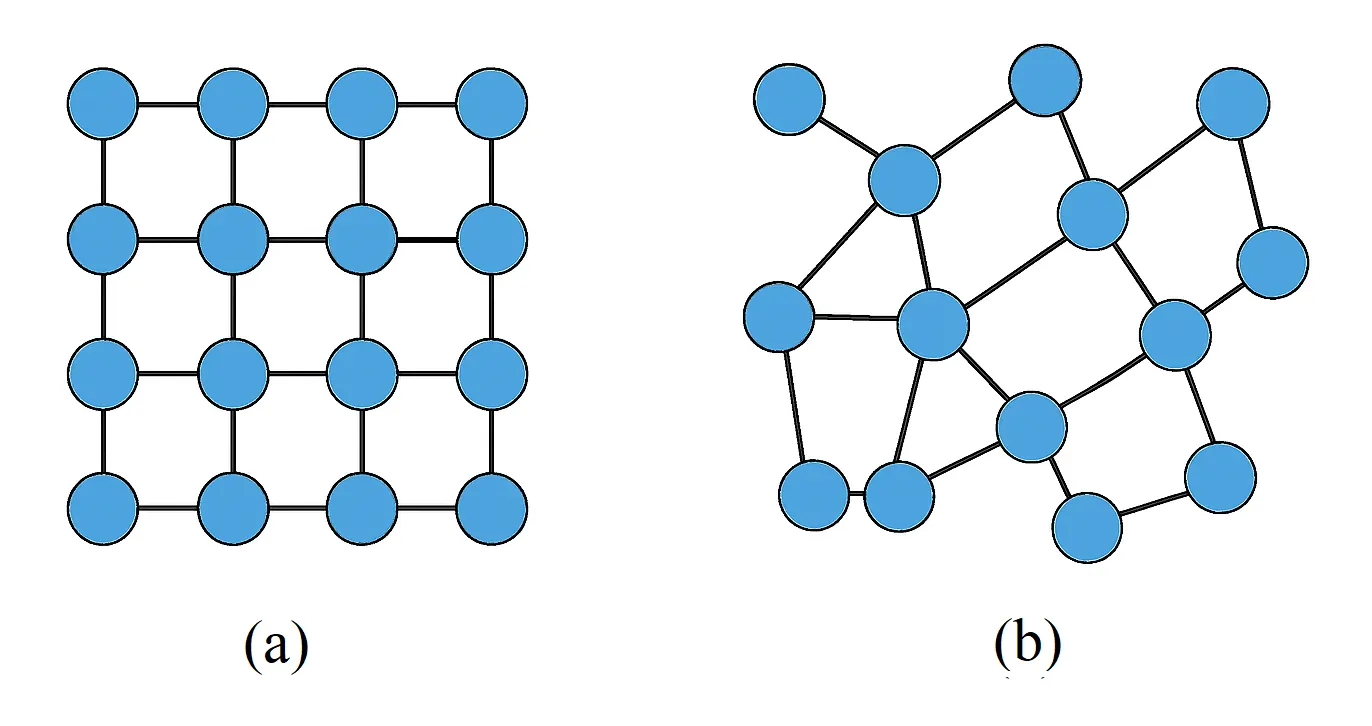
Understanding atomic structures is crucial, yet amorphous materials remain challenging due to their irregular and non-periodic nature. The wavelet-transform radial distribution function (WT-RDF) offers a physics-based framework for analyzing amorphous structures, reliably predicting the first and second RDF peaks and overall curve trends in both binary Ge 0.25 Se 0.75 and ternary Ag x(Ge 0.25 Se 0.75)100-x (x=5,10,15,20,25) systems. Despite these strengths, WT-RDF shows limitations in amplitude accuracy, which affects quantitative analyses such as coordination numbers. This study addresses the issue by optimizing WT-RDF parameters using a machine learning approach, producing the enhanced WT-RDF+ framework. WT-RDF+ improves the precision of peak predictions and outperforms benchmark ML models, including RBF and LSTM, even when trained on only 25 percent of the binary dataset. These results demonstrate that WT-RDF+ is a robust and reliable model for structural characterization of amorphous materials, particularly Ge-Se systems, and support the efficient design and development of phase-change thin films for next-generation electronic devices and components.
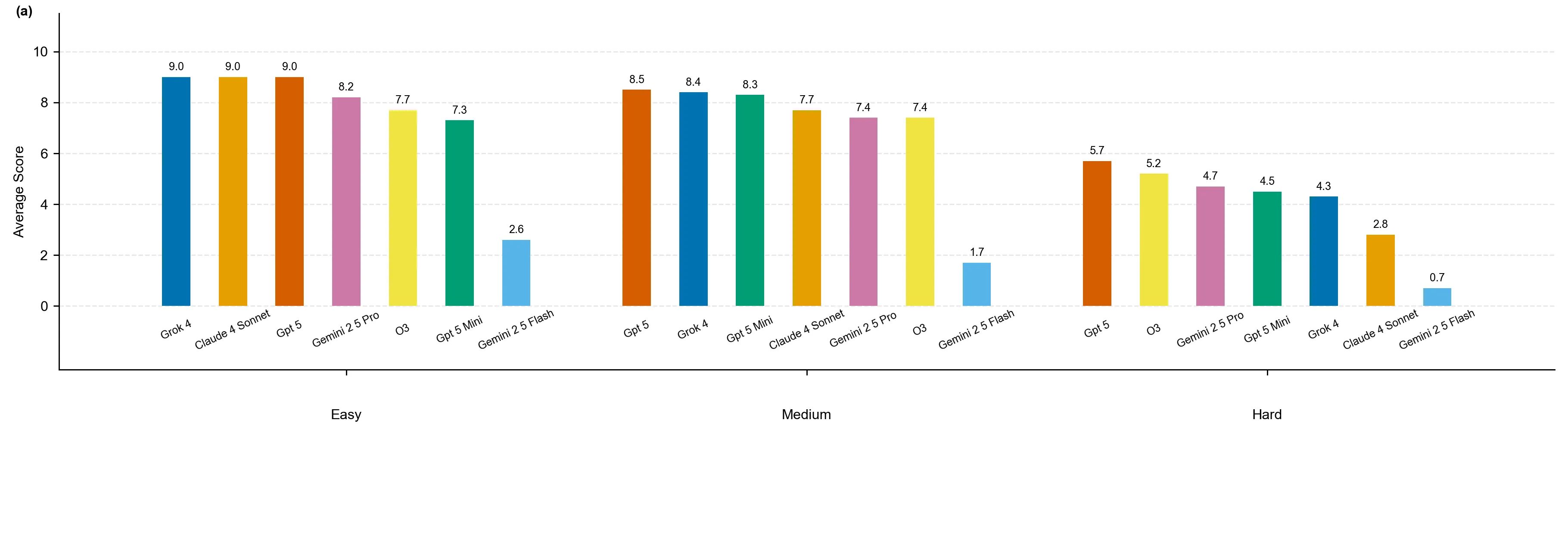
We introduce QMBench, a comprehensive benchmark designed to evaluate the capability of large language model agents in quantum materials research. This specialized benchmark assesses the model's ability to apply condensed matter physics knowledge and computational techniques such as density functional theory to solve research problems in quantum materials science. QMBench encompasses different domains of the quantum material research, including structural properties, electronic properties, thermodynamic and other properties, symmetry principle and computational methodologies. By providing a standardized evaluation framework, QMBench aims to accelerate the development of an AI scientist capable of making creative contributions to quantum materials research. We expect QMBench to be developed and constantly improved by the research community.
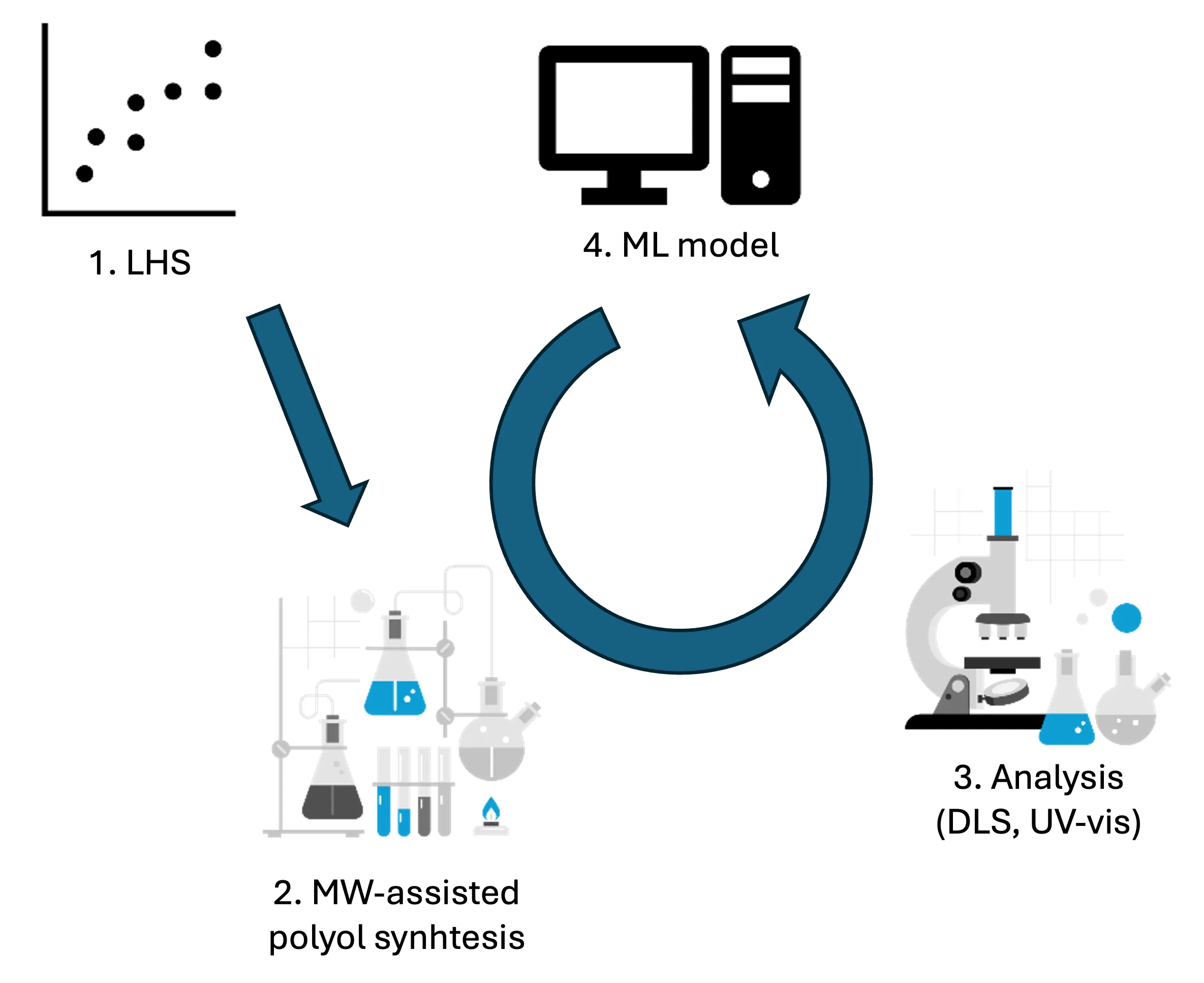
Copper nanoparticles (Cu NPs) have a broad applicability, yet their synthesis is sensitive to subtle changes in reaction parameters. This sensitivity, combined with the time- and resource-intensive nature of experimental optimization, poses a major challenge in achieving reproducible and size-controlled synthesis. While Machine Learning (ML) shows promise in materials research, its application is often limited by scarcity of large high-quality experimental data sets. This study explores ML to predict the size of Cu NPs from microwave-assisted polyol synthesis using a small data set of 25 in-house performed syntheses. Latin Hypercube Sampling is used to efficiently cover the parameter space while creating the experimental data set. Ensemble regression models, built with the AMADEUS framework, successfully predict particle sizes with high accuracy ($R^2 = 0.74$), outperforming classical statistical approaches ($R^2 = 0.60$). Overall, this study highlights that, for lab-scale synthesis optimization, high-quality small datasets combined with classical, interpretable ML models outperform traditional statistical methods and are fully sufficient for quantitative synthesis prediction. This approach provides a sustainable and experimentally realistic pathway toward data-driven inorganic synthesis design.

We develop a general theoretical framework for computing the time-resolved magneto-optical Kerr effect in ultrafast pump-probe setups, formulated within the Dynamical Projective Operatorial Approach (DPOA) and its application to the generalized linear-response theory for pumped systems. Furthermore, we exploit this formalism to express the post-pump optical conductivity and consequently the Kerr rotation in terms of the time-evolved single-particle density matrix (SPDM), providing a transparent and computationally efficient description of photo-excited multi-band systems. This extension, in addition to its lower computational cost, has the advantage of allowing the inclusion of phenomenological damping. We illustrate the formalism using both (i) a two-band tight-binding model, which captures the essential physics of ultrafast spin-charge dynamics and the Kerr rotation, and (ii) weakly spin-polarized germanium, as a realistic playground with a complex band structure. The results demonstrate that, by exploiting DPOA and/or its SPDM extension, one can reliably reproduce both the short-time features under the pump-pulse envelope and the long-time dynamics after excitation, offering a versatile framework for analyzing time-resolved magneto-optical Kerr effect experiments in complex materials. Moreover, this analysis clearly shows that the Kerr rotation can be used to deduce experimentally the relevant n-photon resonances for a given specific material.
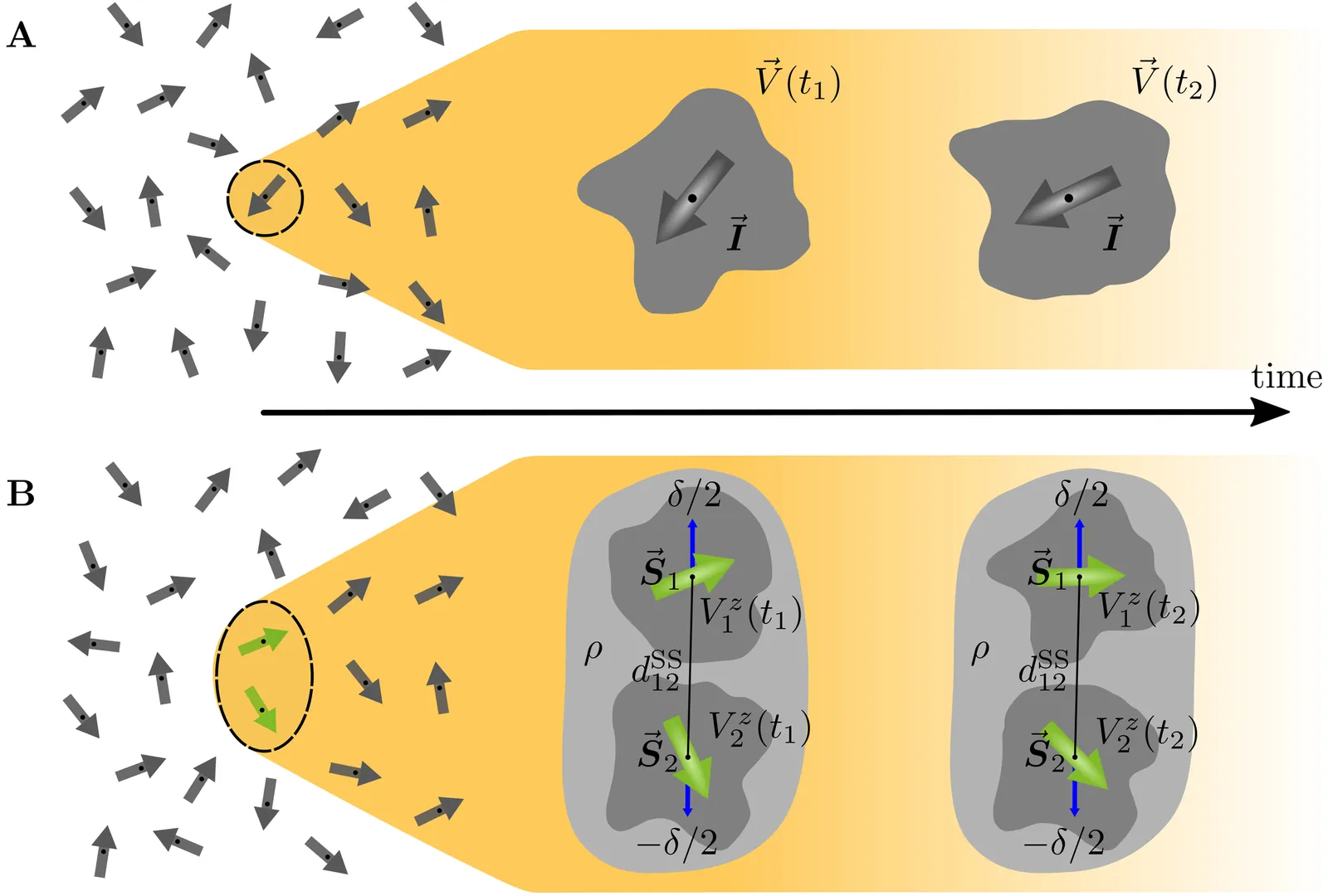
The dynamics of disordered nuclear spin ensembles are the subject of nuclear magnetic resonance studies. Due to the through-space long-range dipolar interaction generically many spins are involved in the time evolution, so that exact brute force calculations are impossible. The recently established spin dynamic mean-field theory (spinDMFT) represents an efficient and unbiased alternative to overcome this challenge. The approach only requires the dipolar couplings as input and the only prerequisite for its applicability is that each spin interacts with a large number of other spins. In this article, we show that spinDMFT can be used to describe spectral spin diffusion in static samples and to simulate zero-quantum line shapes which eluded an efficient quantitative simulation so far to the best of our knowledge. We perform benchmarks for two test substances that establish an excellent match with published experimental data. As spinDMFT combines low computational effort with high accuracy, we strongly suggest to use it for large-scale simulations of spin diffusion, which are important in various areas of magnetic resonance.
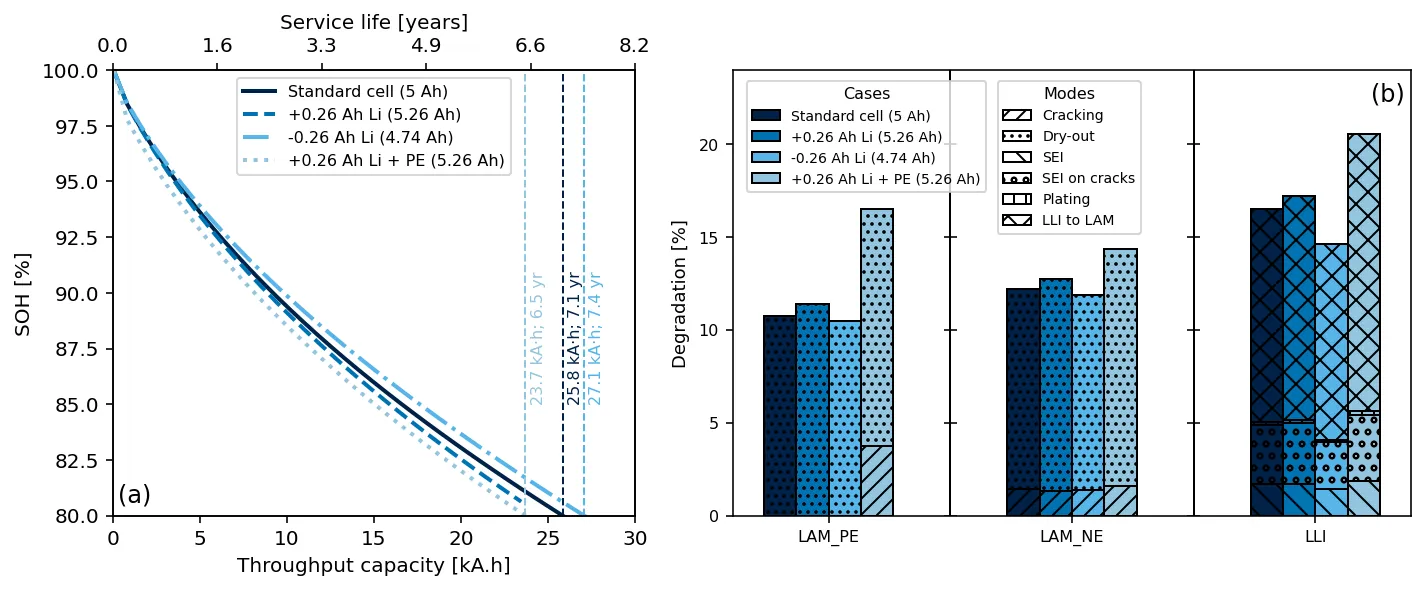
Designing lithium-ion batteries for long service life remains a challenge, as most cells are optimized for beginning-of-life metrics such as energy density, often overlooking how design and operating conditions shape degradation. This work introduces a degradation-aware design framework built around finite, interacting reservoirs (lithium, porosity, and electrolyte) that are depleted over time by coupled degradation processes. We extend a physics-based Doyle-Fuller-Newman model to include validated mechanisms such as SEI growth, lithium plating, cracking, and solvent dry-out, and simulate how small design changes impact lifetime. Across more than 1,000 cycles, we find that increasing electrolyte volume by just 1% or porosity by 5% can extend service life by over 30% without significantly affecting cell energy density. However, lithium excess, while boosting initial capacity, can accelerate failure if not supported by sufficient structural or ionic buffers. Importantly, we show that interaction between reservoirs is crucial to optimal design: multi-reservoir tuning yields either synergistic benefits or compound failures, depending on operating conditions. We also quantify how C-rate and operating temperature influence degradation pathways, emphasizing the need for co-optimized design and usage profiles. By reframing degradation as a problem of managing finite internal reservoirs, this work offers a predictive and mechanistic foundation for designing lithium-ion batteries that balance energy, durability, and application-specific needs.