Biomolecules
arXiv:q-bio.BM
DNA, RNA, proteins, lipids; molecular structures and dynamics; enzymes, membranes.
Looking for a broader view? This category is part of:
DNA, RNA, proteins, lipids; molecular structures and dynamics; enzymes, membranes.
Looking for a broader view? This category is part of:
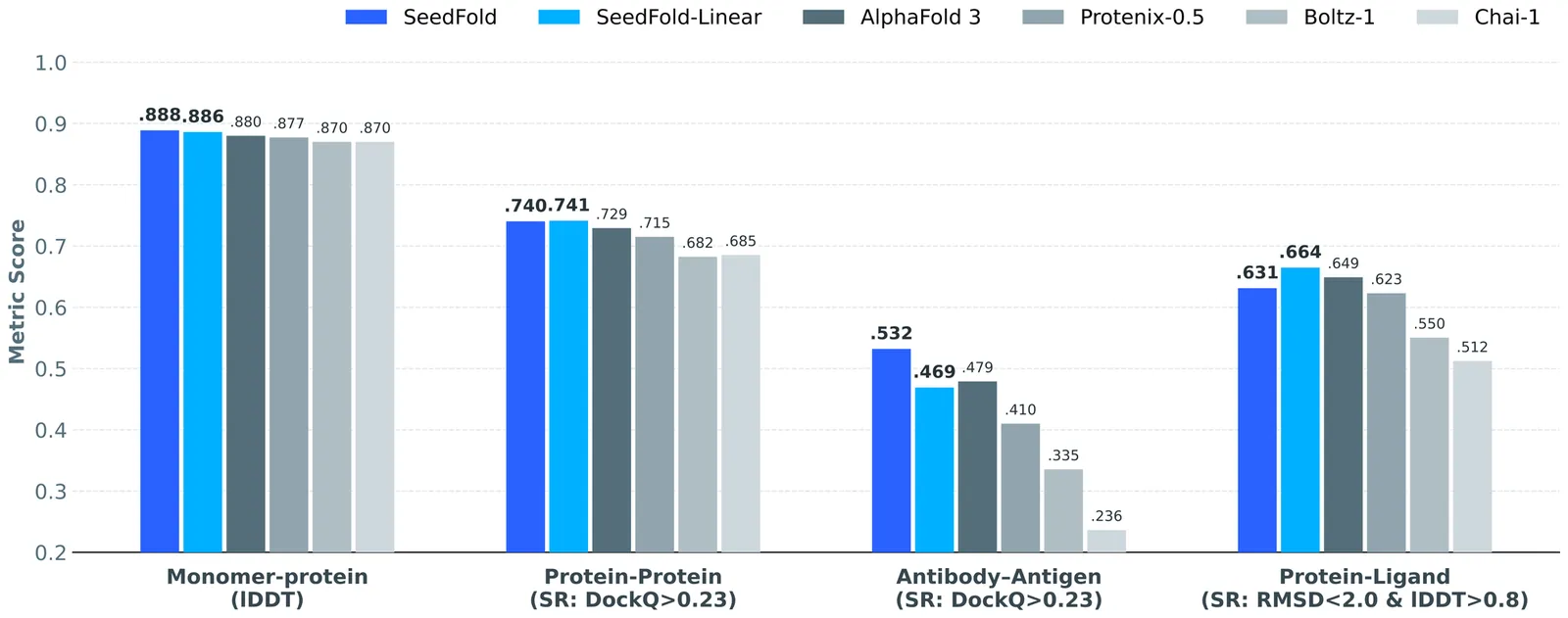
Highly accurate biomolecular structure prediction is a key component of developing biomolecular foundation models, and one of the most critical aspects of building foundation models is identifying the recipes for scaling the model. In this work, we present SeedFold, a folding model that successfully scales up the model capacity. Our contributions are threefold: first, we identify an effective width-scaling strategy for the Pairformer to increase representation capacity; second, we introduce a novel linear triangular attention that reduces computational complexity to enable efficient scaling; finally, we construct a large-scale distillation dataset to substantially enlarge the training set. Experiments on FoldBench show that SeedFold outperforms AlphaFold3 on most protein-related tasks.
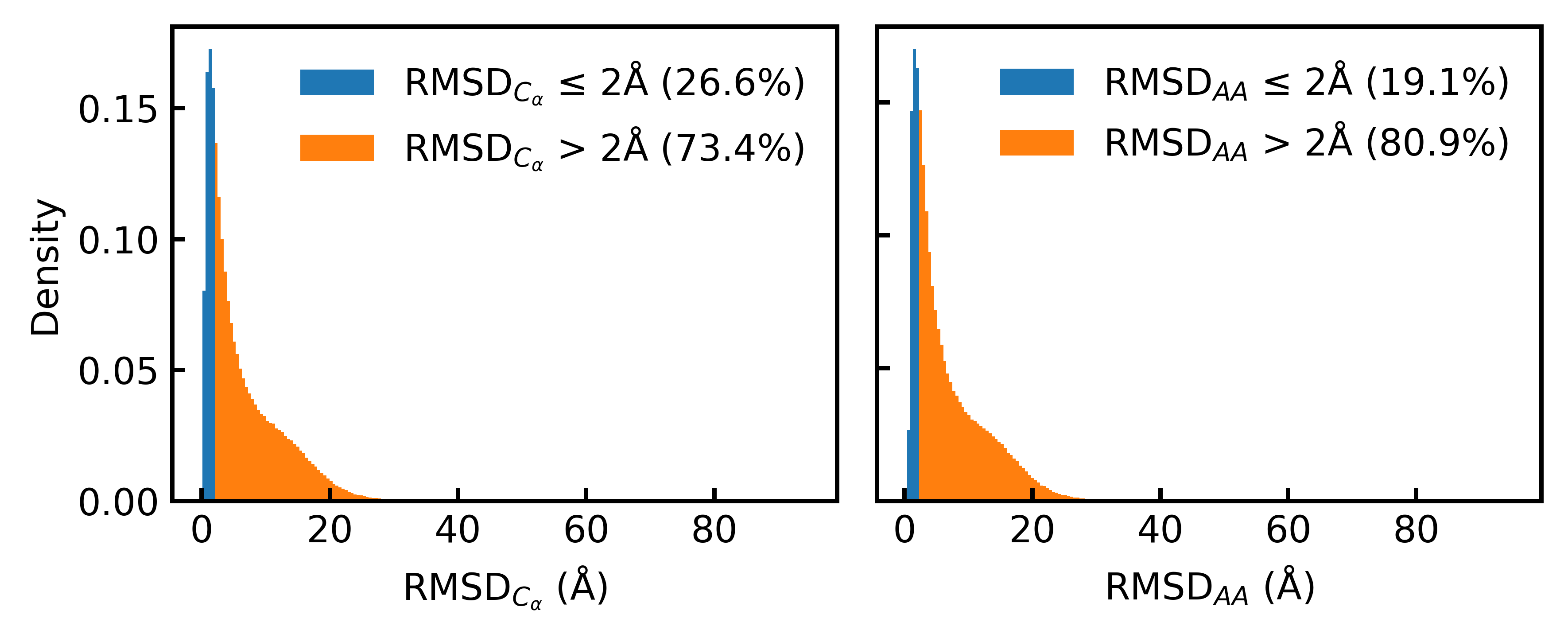
High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteina, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteina Atomistica, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving \method's structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.
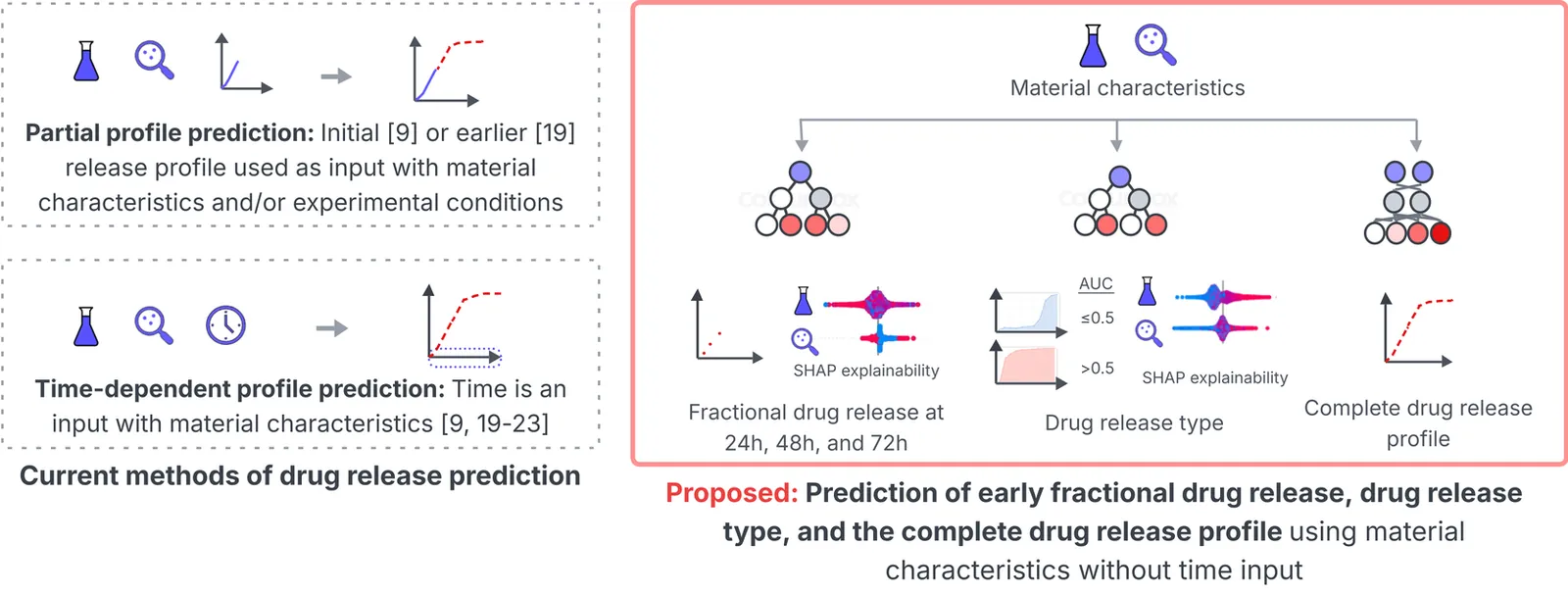
Polymer-based long-acting injectables (LAIs) have transformed the treatment of chronic diseases by enabling controlled drug delivery, thus reducing dosing frequency and extending therapeutic duration. Achieving controlled drug release from LAIs requires extensive optimization of the complex underlying physicochemical properties. Machine learning (ML) can accelerate LAI development by modeling the complex relationships between LAI properties and drug release. However, recent ML studies have provided limited information on key properties that modulate drug release, due to the lack of custom modeling and analysis tailored to LAI data. This paper presents a novel data transformation and explainable ML approach to synthesize actionable information from 321 LAI formulations by predicting early drug release at 24, 48, and 72 hours, classification of release profile types, and prediction of complete release profiles. These three experiments investigate the contribution and control of LAI material characteristics in early and complete drug release profiles. A strong correlation (>0.65) is observed between the true and predicted drug release in 72 hours, while a 0.87 F1-score is obtained in classifying release profile types. A time-independent ML framework predicts delayed biphasic and triphasic curves with better performance than current time-dependent approaches. Shapley additive explanations reveal the relative influence of material characteristics during early and for complete release which fill several gaps in previous in-vitro and ML-based studies. The novel approach and findings can provide a quantitative strategy and recommendations for scientists to optimize the drug-release dynamics of LAI. The source code for the model implementation is publicly available.
Highly accurate biomolecular structure prediction is a key component of developing biomolecular foundation models, and one of the most critical aspects of building foundation models is identifying the recipes for scaling the model. In this work, we present SeedFold, a folding model that successfully scales up the model capacity. Our contributions are threefold: first, we identify an effective width-scaling strategy for the Pairformer to increase representation capacity; second, we introduce a novel linear triangular attention that reduces computational complexity to enable efficient scaling; finally, we construct a large-scale distillation dataset to substantially enlarge the training set. Experiments on FoldBench show that SeedFold outperforms AlphaFold3 on most protein-related tasks.

Can machine learning models identify which chemist made a molecule from structure alone? If so, models trained on literature data may exploit chemist intent rather than learning causal structure-activity relationships. We test this by linking CHEMBL assays to publication authors and training a 1,815-class classifier to predict authors from molecular fingerprints, achieving 60% top-5 accuracy under scaffold-based splitting. We then train an activity model that receives only a protein identifier and an author-probability vector derived from structure, with no direct access to molecular descriptors. This author-only model achieves predictive power comparable to a simple baseline that has access to structure. This reveals a "Clever Hans" failure mode: models can predict bioactivity largely by inferring chemist goals and favorite targets without requiring a lab-independent understanding of chemistry. We analyze the sources of this leakage, propose author-disjoint splits, and recommend dataset practices to decouple chemist intent from biological outcomes.
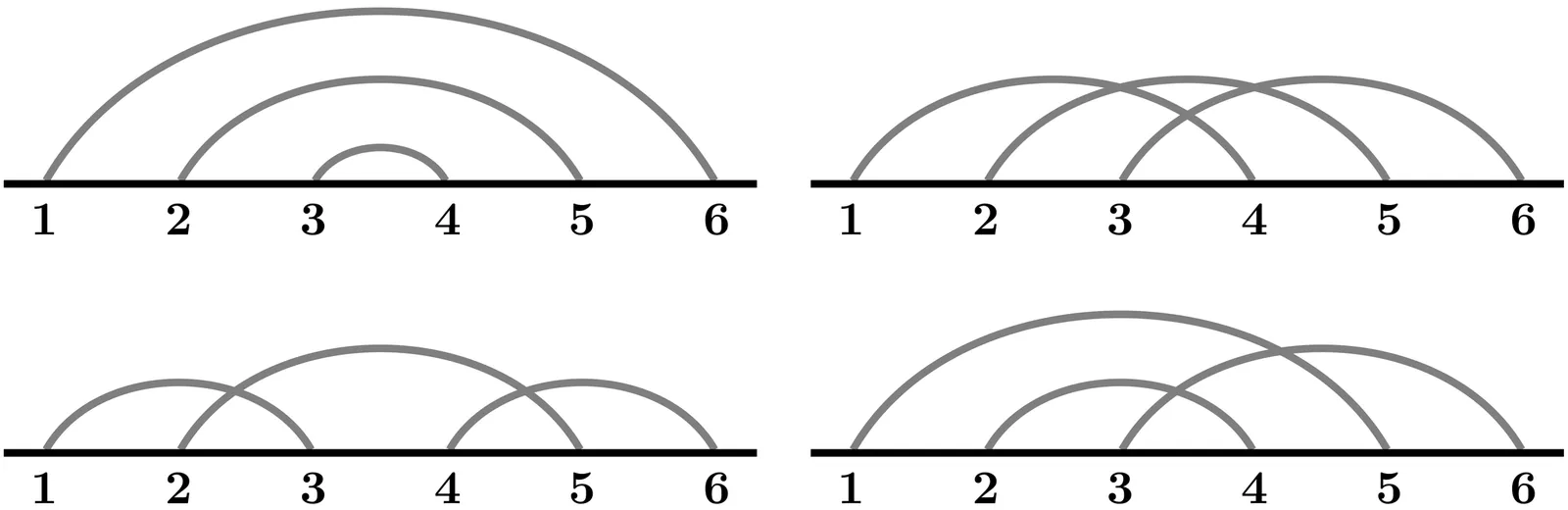
RNA molecules are known to form complex secondary structures including pseudoknots. A systematic framework for the enumeration, classification and prediction of secondary structures is critical to determine the biological significance of the molecular configurations of RNA. Chord diagrams are mathematical objects widely used to represent RNA secondary structures and to analyze structural motifs, however a mathematically rigorous enumeration of pseudoknots remains a challenge. We introduce a method that incorporates a distance-based metric $τ$ to analyze the intersection graph of a chord diagram associated with a pseudoknotted structure. In particular, our method formally defines a pseudoknot in terms of a weighted vertex cover of a certain intersection graph constructed from a partition of the chord diagram representing the nucleotide sequence of the RNA molecule. In this graph-theoretic context, we introduce a rigorous algorithm that enumerates pseudoknots, classifies secondary structures, and is sensitive to three-dimensional topological features. We implement our methods in MATLAB and test the algorithm on pseudoknotted structures from the bpRNA-1m database. Our findings confirm that genus is a robust quantifier of pseudoknot complexity.
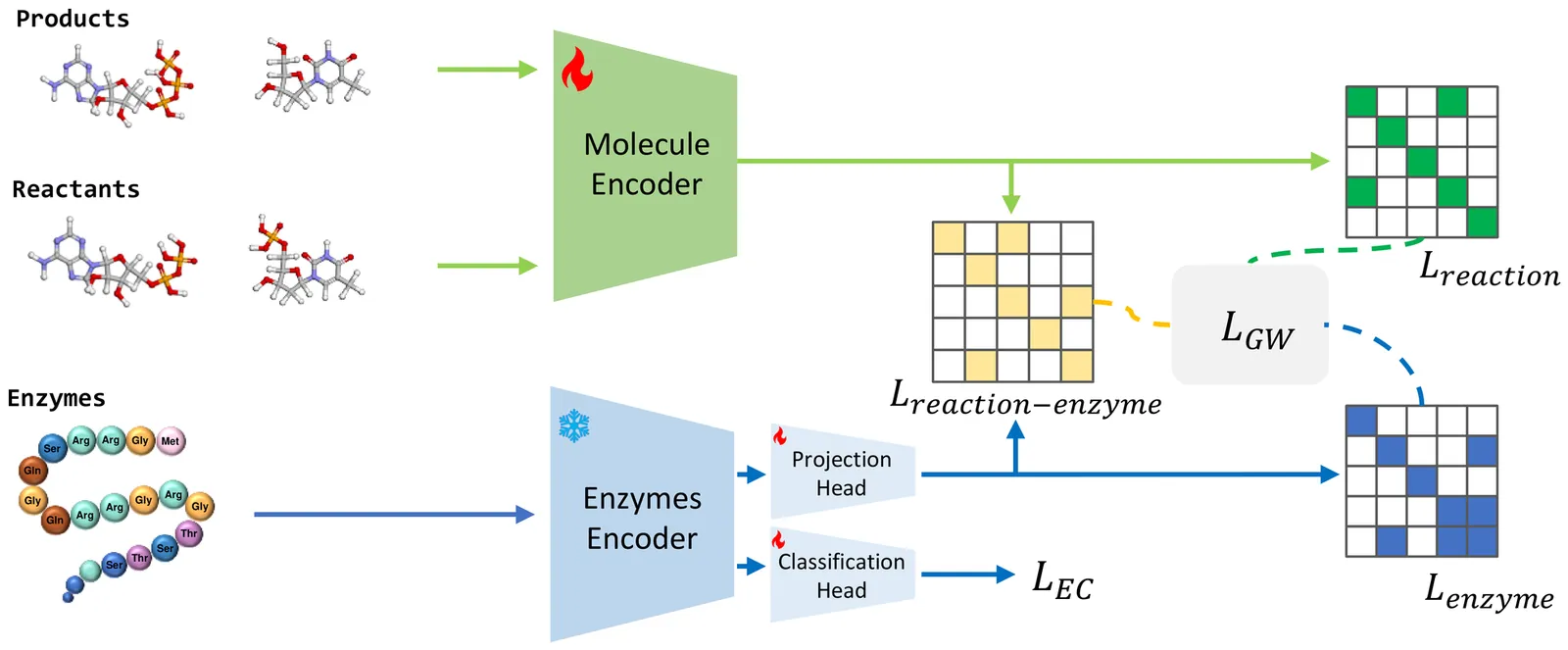
Enzymes are crucial catalysts that enable a wide range of biochemical reactions. Efficiently identifying specific enzymes from vast protein libraries is essential for advancing biocatalysis. Traditional computational methods for enzyme screening and retrieval are time-consuming and resource-intensive. Recently, deep learning approaches have shown promise. However, these methods focus solely on the interaction between enzymes and reactions, overlooking the inherent hierarchical relationships within each domain. To address these limitations, we introduce FGW-CLIP, a novel contrastive learning framework based on optimizing the fused Gromov-Wasserstein distance. FGW-CLIP incorporates multiple alignments, including inter-domain alignment between reactions and enzymes and intra-domain alignment within enzymes and reactions. By introducing a tailored regularization term, our method minimizes the Gromov-Wasserstein distance between enzyme and reaction spaces, which enhances information integration across these domains. Extensive evaluations demonstrate the superiority of FGW-CLIP in challenging enzyme-reaction tasks. On the widely-used EnzymeMap benchmark, FGW-CLIP achieves state-of-the-art performance in enzyme virtual screening, as measured by BEDROC and EF metrics. Moreover, FGW-CLIP consistently outperforms across all three splits of ReactZyme, the largest enzyme-reaction benchmark, demonstrating robust generalization to novel enzymes and reactions. These results position FGW-CLIP as a promising framework for enzyme discovery in complex biochemical settings, with strong adaptability across diverse screening scenarios.
Understanding how protein mutations affect protein structure is essential for advancements in computational biology and bioinformatics. We introduce PRIMRose, a novel approach that predicts energy values for each residue given a mutated protein sequence. Unlike previous models that assess global energy shifts, our method analyzes the localized energetic impact of double amino acid insertions or deletions (InDels) at the individual residue level, enabling residue-specific insights into structural and functional disruption. We implement a Convolutional Neural Network architecture to predict the energy changes of each residue in a protein mutation. We train our model on datasets constructed from nine proteins, grouped into three categories: one set with exhaustive double InDel mutations, another with approximately 145k randomly sampled double InDel mutations, and a third with approximately 80k randomly sampled double InDel mutations. Our model achieves high predictive accuracy across a range of energy metrics as calculated by the Rosetta molecular modeling suite and reveals localized patterns that influence model performance, such as solvent accessibility and secondary structure context. This per-residue analysis offers new insights into the mutational tolerance of specific regions within proteins and provides higher interpretable and biologically meaningful predictions of InDels' effects.
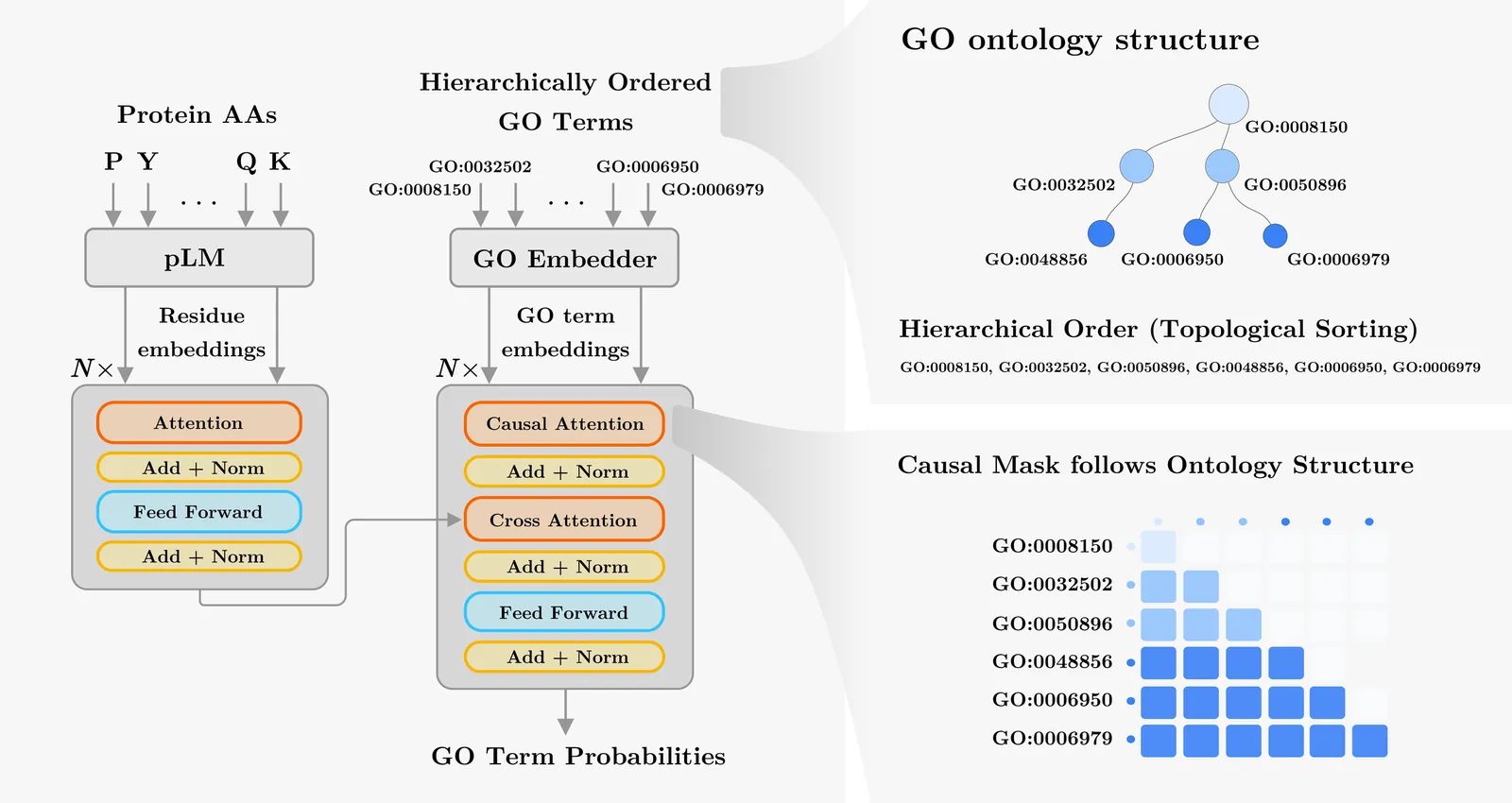
Accurate prediction of protein function is essential for elucidating molecular mechanisms and advancing biological and therapeutic discovery. Yet experimental annotation lags far behind the rapid growth of protein sequence data. Computational approaches address this gap by associating proteins with Gene Ontology (GO) terms, which encode functional knowledge through hierarchical relations and textual definitions. However, existing models often emphasize one modality over the other, limiting their ability to generalize, particularly to unseen or newly introduced GO terms that frequently arise as the ontology evolves, and making the previously trained models outdated. We present STAR-GO, a Transformer-based framework that jointly models the semantic and structural characteristics of GO terms to enhance zero-shot protein function prediction. STAR-GO integrates textual definitions with ontology graph structure to learn unified GO representations, which are processed in hierarchical order to propagate information from general to specific terms. These representations are then aligned with protein sequence embeddings to capture sequence-function relationships. STAR-GO achieves state-of-the-art performance and superior zero-shot generalization, demonstrating the utility of integrating semantics and structure for robust and adaptable protein function prediction. Code is available at https://github.com/boun-tabi-lifelu/stargo.
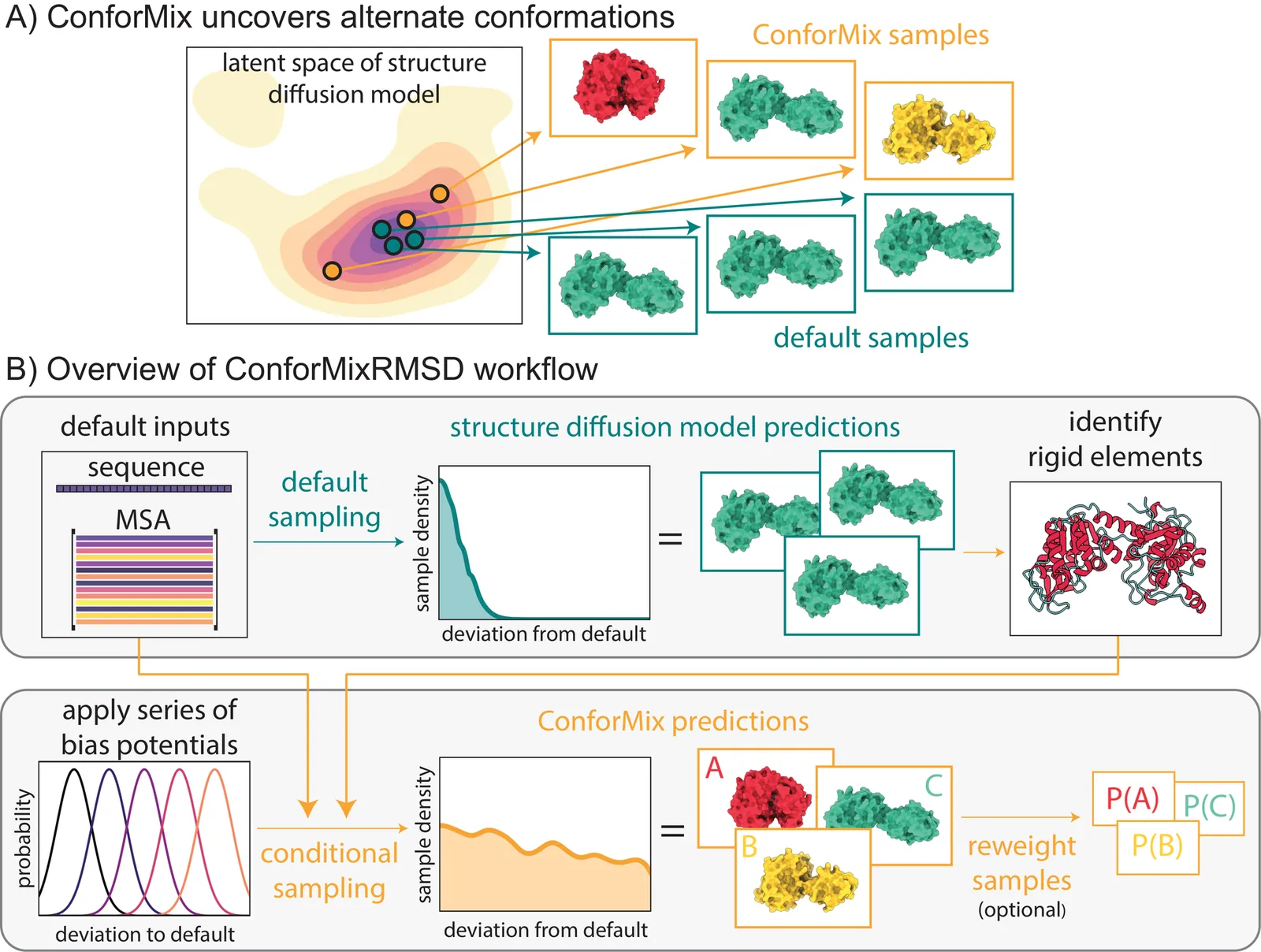
The function of biomolecules such as proteins depends on their ability to interconvert between a wide range of structures or "conformations." Researchers have endeavored for decades to develop computational methods to predict the distribution of conformations, which is far harder to determine experimentally than a static folded structure. We present ConforMix, an inference-time algorithm that enhances sampling of conformational distributions using a combination of classifier guidance, filtering, and free energy estimation. Our approach upgrades diffusion models -- whether trained for static structure prediction or conformational generation -- to enable more efficient discovery of conformational variability without requiring prior knowledge of major degrees of freedom. ConforMix is orthogonal to improvements in model pretraining and would benefit even a hypothetical model that perfectly reproduced the Boltzmann distribution. Remarkably, when applied to a diffusion model trained for static structure prediction, ConforMix captures structural changes including domain motion, cryptic pocket flexibility, and transporter cycling, while avoiding unphysical states. Case studies of biologically critical proteins demonstrate the scalability, accuracy, and utility of this method.
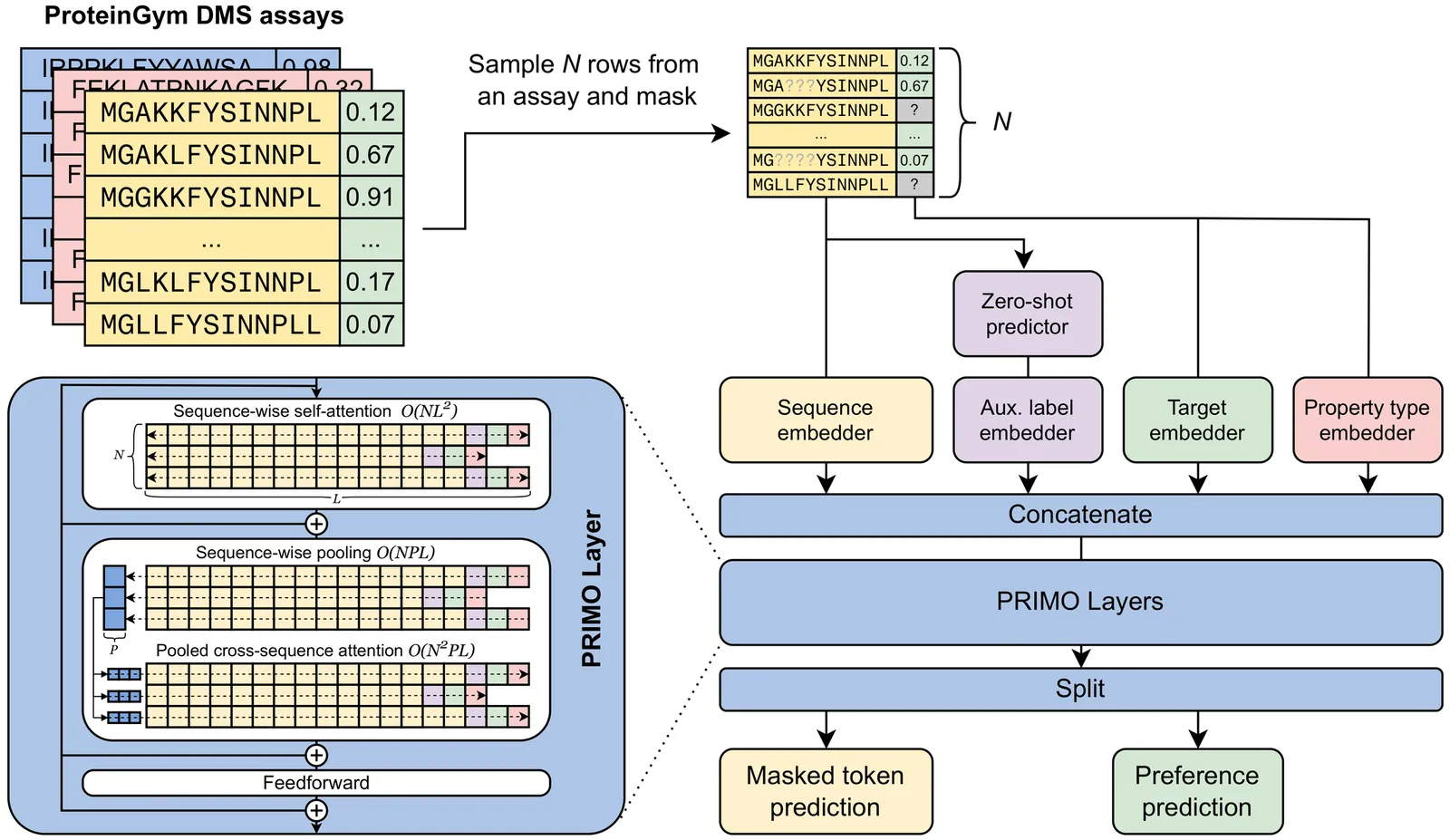
Accurately predicting protein fitness with minimal experimental data is a persistent challenge in protein engineering. We introduce PRIMO (PRotein In-context Mutation Oracle), a transformer-based framework that leverages in-context learning and test-time training to adapt rapidly to new proteins and assays without large task-specific datasets. By encoding sequence information, auxiliary zero-shot predictions, and sparse experimental labels from many assays as a unified token set in a pre-training masked-language modeling paradigm, PRIMO learns to prioritize promising variants through a preference-based loss function. Across diverse protein families and properties-including both substitution and indel mutations-PRIMO outperforms zero-shot and fully supervised baselines. This work underscores the power of combining large-scale pre-training with efficient test-time adaptation to tackle challenging protein design tasks where data collection is expensive and label availability is limited.
High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteina, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteina Atomistica, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving \method's structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.

Accurate prediction of enzyme kinetic parameters is crucial for drug discovery, metabolic engineering, and synthetic biology applications. Current computational approaches face limitations in capturing complex enzyme-substrate interactions and often focus on single parameters while neglecting the joint prediction of catalytic turnover numbers (Kcat) and Michaelis-Menten constants (Km). We present EnzyCLIP, a novel dual-encoder framework that leverages contrastive learning and cross-attention mechanisms to predict enzyme kinetic parameters from protein sequences and substrate molecular structures. Our approach integrates ESM-2 protein language model embeddings with ChemBERTa chemical representations through a CLIP-inspired architecture enhanced with bidirectional cross-attention for dynamic enzyme-substrate interaction modeling. EnzyCLIP combines InfoNCE contrastive loss with Huber regression loss to learn aligned multimodal representations while predicting log10-transformed kinetic parameters. The model is trained on the CatPred-DB database containing 23,151 Kcat and 41,174 Km experimentally validated measurements, and achieved competitive performance with R2 scores of 0.593 for Kcat and 0.607 for Km prediction. XGBoost ensemble methods applied to the learned embeddings further improved Km prediction (R2 = 0.61) while maintaining robust Kcat performance.
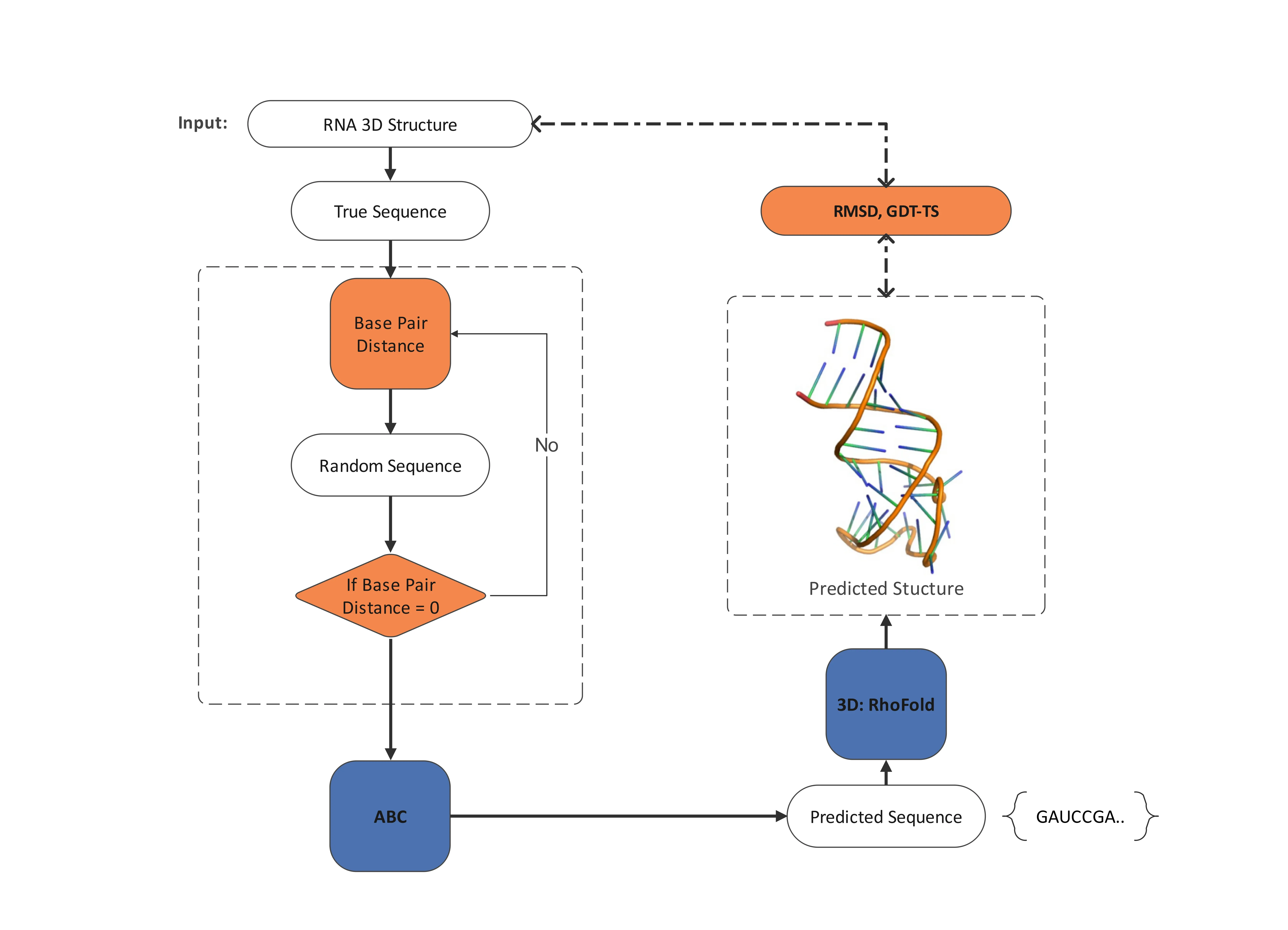
The Ribonucleic Acid (RNA) inverse folding problem, designing nucleotide sequences that fold into specific tertiary structures, is a fundamental computational biology problem with important applications in synthetic biology and bioengineering. The design of complex three-dimensional RNA architectures remains computationally demanding and mostly unresolved, as most existing approaches focus on secondary structures. In order to address tertiary RNA inverse folding, we present BeeRNA, a bio-inspired method that employs the Artificial Bee Colony (ABC) optimization algorithm. Our approach combines base-pair distance filtering with RMSD-based structural assessment using RhoFold for structure prediction, resulting in a two-stage fitness evaluation strategy. To guarantee biologically plausible sequences with balanced GC content, the algorithm takes thermodynamic constraints and adaptive mutation rates into consideration. In this work, we focus primarily on short and medium-length RNAs ($<$ 100 nucleotides), a biologically significant regime that includes microRNAs (miRNAs), aptamers, and ribozymes, where BeeRNA achieves high structural fidelity with practical CPU runtimes. The lightweight, training-free implementation will be publicly released for reproducibility, offering a promising bio-inspired approach for RNA design in therapeutics and biotechnology.

Designing new protein structures is fundamental to computational biology, enabling advances in therapeutic molecule discovery and enzyme engineering. Existing diffusion-based generative models typically operate in Cartesian coordinate space, where adding noise disrupts strict geometric constraints such as fixed bond lengths and angles, often producing physically invalid structures. To address this limitation, we propose a Torsion-Space Diffusion Model that generates protein backbones by denoising torsion angles, ensuring perfect local geometry by construction. A differentiable forward-kinematics module reconstructs 3D coordinates with fixed 3.8 Angstrom backbone bond lengths while a constrained post-processing refinement optimizes global compactness via Radius of Gyration (Rg) correction, without violating bond constraints. Experiments on standard PDB proteins demonstrate 100% bond-length accuracy and significantly improved structural compactness, reducing Rg error from 70% to 18.6% compared to Cartesian diffusion baselines. Overall, this hybrid torsion-diffusion plus geometric-refinement framework generates physically valid and compact protein backbones, providing a promising path toward full-atom protein generation.
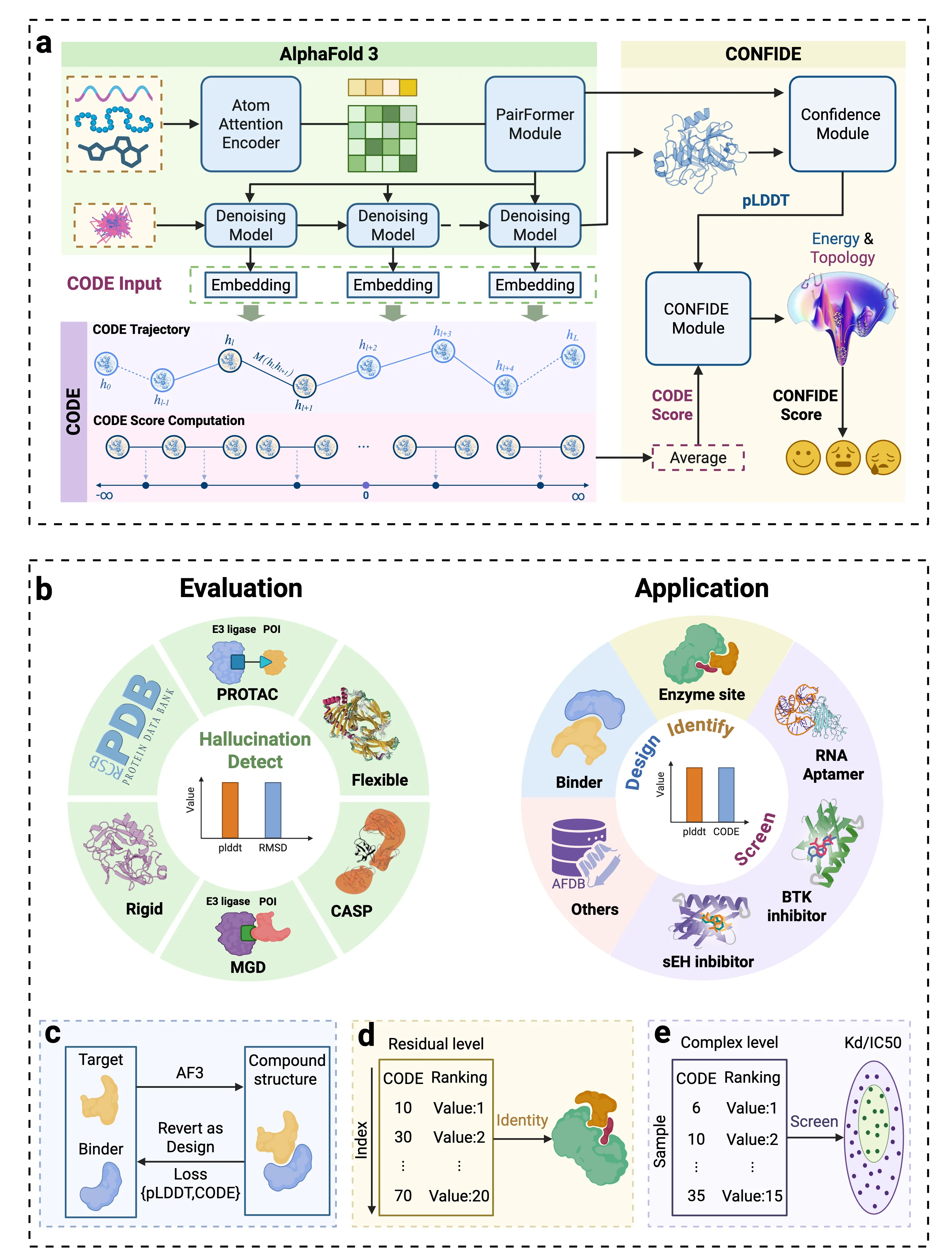
Reliable evaluation of protein structure predictions remains challenging, as metrics like pLDDT capture energetic stability but often miss subtle errors such as atomic clashes or conformational traps reflecting topological frustration within the protein folding energy landscape. We present CODE (Chain of Diffusion Embeddings), a self evaluating metric empirically found to quantify topological frustration directly from the latent diffusion embeddings of the AlphaFold3 series of structure predictors in a fully unsupervised manner. Integrating this with pLDDT, we propose CONFIDE, a unified evaluation framework that combines energetic and topological perspectives to improve the reliability of AlphaFold3 and related models. CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT's 0.33 (a relative improvement of 148\%). CONFIDE significantly enhances the reliability of quality evaluation in molecular glue structure prediction benchmarks, achieving a Spearman correlation of 0.73 with RMSD, compared to pLDDT's correlation of 0.42, a relative improvement of 73.8\%. Beyond quality assessment, our approach applies to diverse drug design tasks, including all-atom binder design, enzymatic active site mapping, mutation induced binding affinity prediction, nucleic acid aptamer screening, and flexible protein modeling. By combining data driven embeddings with theoretical insight, CODE and CONFIDE outperform existing metrics across a wide range of biomolecular systems, offering robust and versatile tools to refine structure predictions, advance structural biology, and accelerate drug discovery.
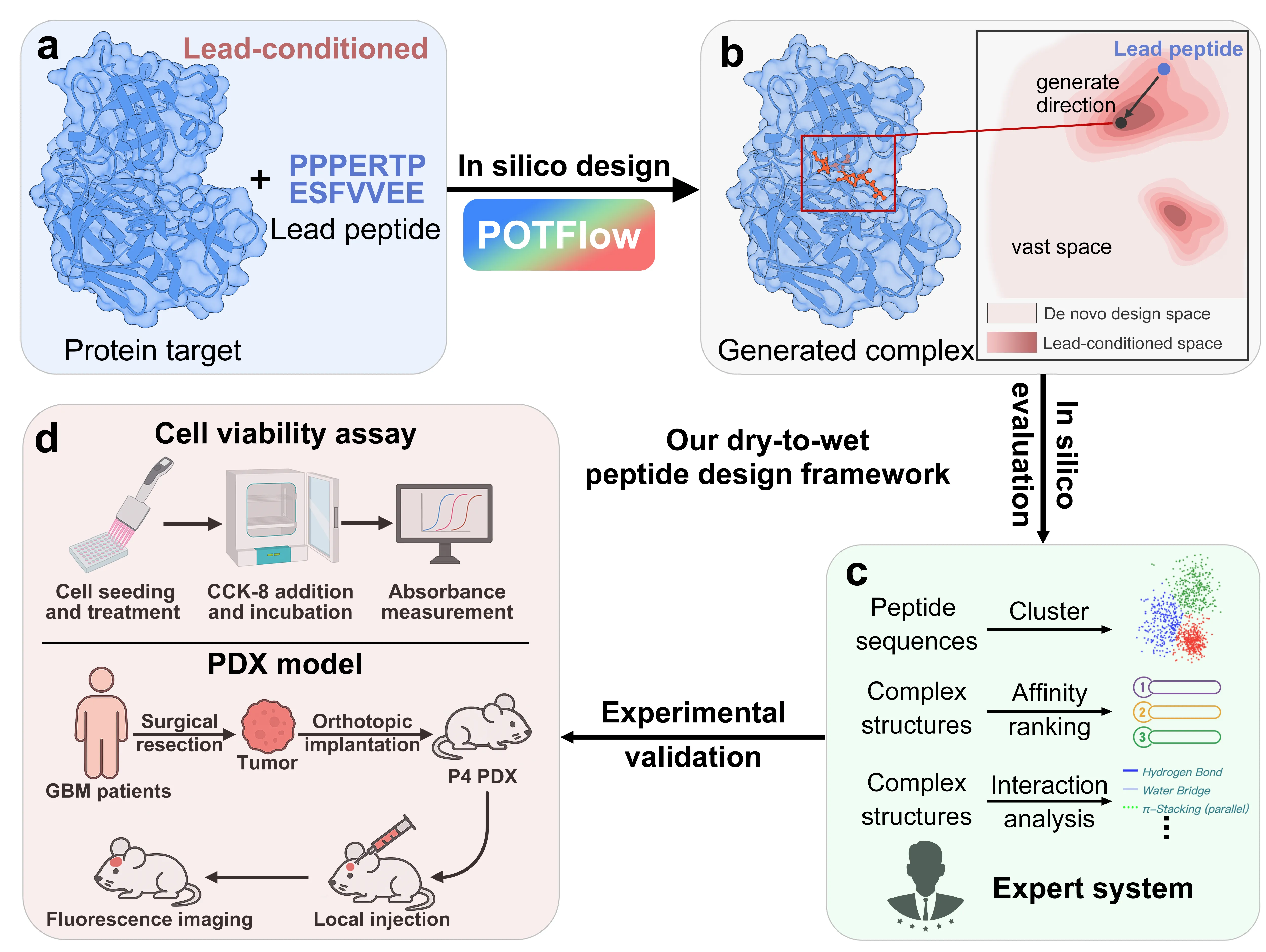
Glioblastoma (GBM) remains the most aggressive tumor, urgently requiring novel therapeutic strategies. Here, we present a dry-to-wet framework combining generative modeling and experimental validation to optimize peptides targeting ATP5A, a potential peptide-binding protein for GBM. Our framework introduces the first lead-conditioned generative model, which focuses exploration on geometrically relevant regions around lead peptides and mitigates the combinatorial complexity of de novo methods. Specifically, we propose POTFlow, a \underline{P}rior and \underline{O}ptimal \underline{T}ransport-based \underline{Flow}-matching model for peptide optimization. POTFlow employs secondary structure information (e.g., helix, sheet, loop) as geometric constraints, which are further refined by optimal transport to produce shorter flow paths. With this design, our method achieves state-of-the-art performance compared with five popular approaches. When applied to GBM, our method generates peptides that selectively inhibit cell viability and significantly prolong survival in a patient-derived xenograft (PDX) model. As the first lead peptide-conditioned flow matching model, POTFlow holds strong potential as a generalizable framework for therapeutic peptide design.
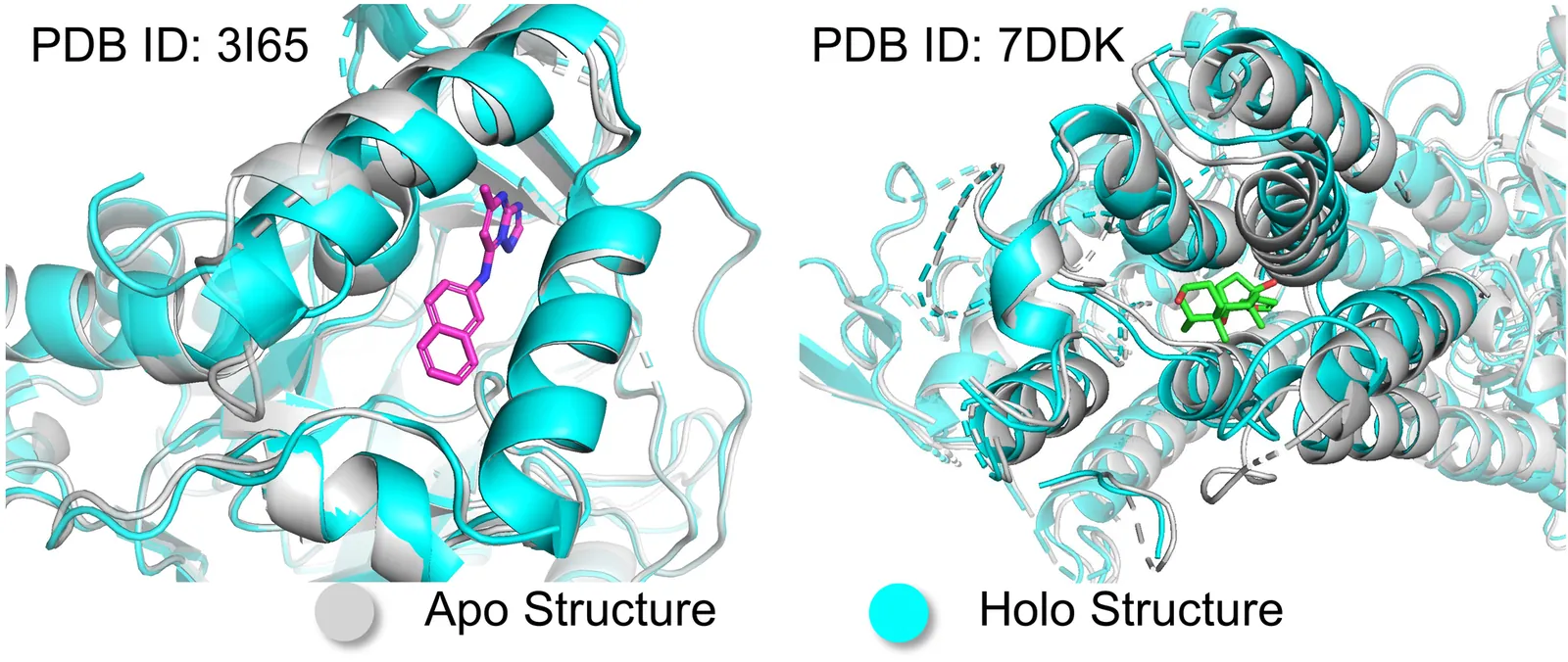
Deep generative models are rapidly advancing structure-based drug design, offering substantial promise for generating small molecule ligands that bind to specific protein targets. However, most current approaches assume a rigid protein binding pocket, neglecting the intrinsic flexibility of proteins and the conformational rearrangements induced by ligand binding, limiting their applicability in practical drug discovery. Here, we propose Apo2Mol, a diffusion-based generative framework for 3D molecule design that explicitly accounts for conformational flexibility in protein binding pockets. To support this, we curate a dataset of over 24,000 experimentally resolved apo-holo structure pairs from the Protein Data Bank, enabling the characterization of protein structure changes associated with ligand binding. Apo2Mol employs a full-atom hierarchical graph-based diffusion model that simultaneously generates 3D ligand molecules and their corresponding holo pocket conformations from input apo states. Empirical studies demonstrate that Apo2Mol can achieve state-of-the-art performance in generating high-affinity ligands and accurately capture realistic protein pocket conformational changes.
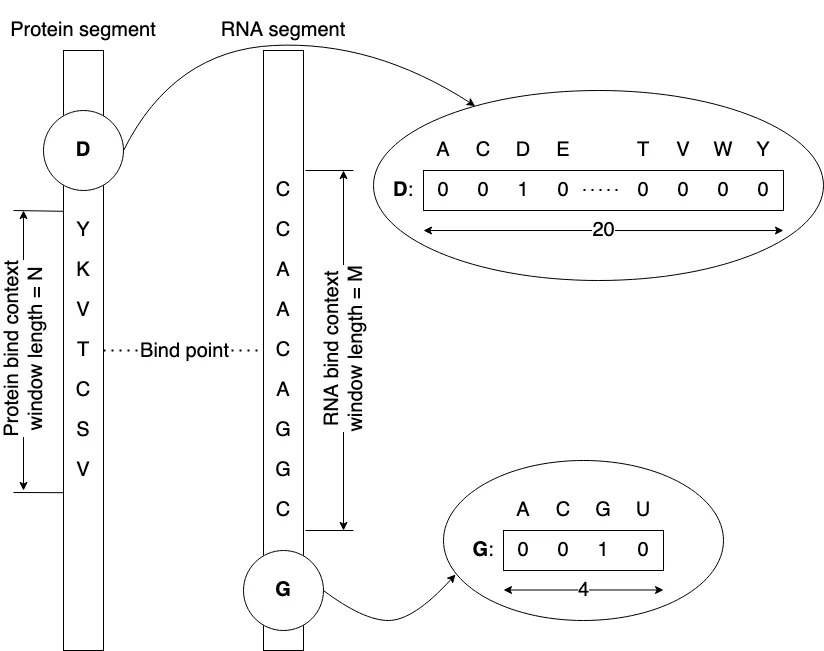
Large Artificial Neural Network (ANN) models have demonstrated success in various domains, including general text and image generation, drug discovery, and protein-RNA (ribonucleic acid) binding tasks. However, these models typically demand substantial computational resources, time, and data for effective training. Given that such extensive resources are often inaccessible to many researchers and that life sciences data sets are frequently limited, we investigated whether small ANN models could achieve acceptable accuracy in protein-RNA prediction. We experimented with shallow feed-forward ANNs comprising two hidden layers and various non-linearities. These models did not utilize explicit structural information; instead, a sliding window approach was employed to implicitly consider the context of neighboring residues and bases. We explored different training techniques to address the issue of highly unbalanced data. Among the seven most popular non-linearities for feed-forward ANNs, only three: Rectified Linear Unit (ReLU), Gated Linear Unit (GLU), and Hyperbolic Tangent (Tanh) yielded converging models. Common re-balancing techniques, such as under- and over-sampling of training sets, proved ineffective, whereas increasing the volume of training data and using model ensembles significantly improved performance. The optimal context window size, balancing both false negative and false positive errors, was found to be approximately 30 residues and bases. Our findings indicate that high-accuracy protein-RNA binding prediction is achievable using computing hardware accessible to most educational and research institutions.
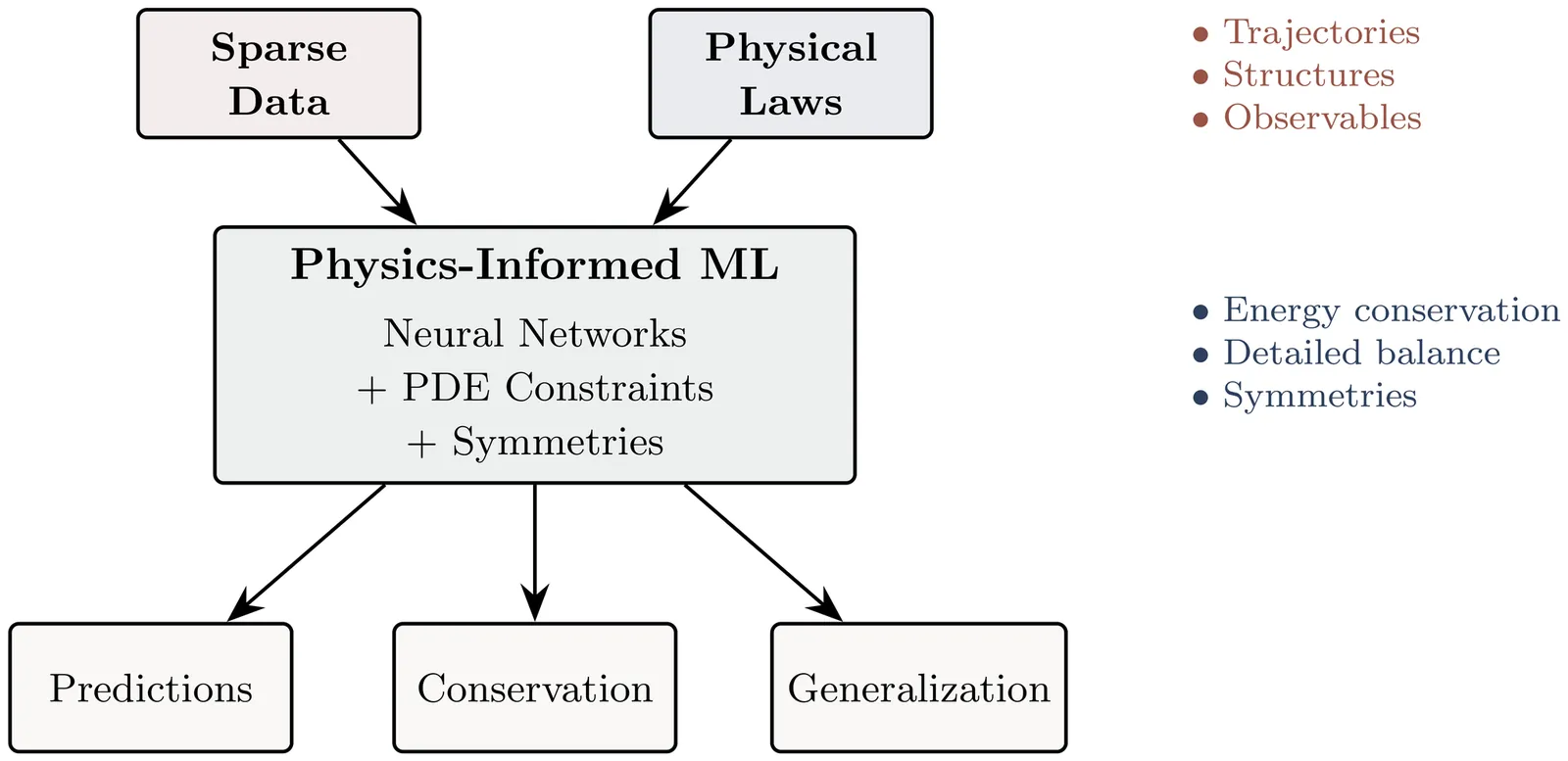
The convergence of statistical learning and molecular physics is transforming our approach to modeling biomolecular systems. Physics-informed machine learning (PIML) offers a systematic framework that integrates data-driven inference with physical constraints, resulting in models that are accurate, mechanistic, generalizable, and able to extrapolate beyond observed domains. This review surveys recent advances in physics-informed neural networks and operator learning, differentiable molecular simulation, and hybrid physics-ML potentials, with emphasis on long-timescale kinetics, rare events, and free-energy estimation. We frame these approaches as solutions to the "biomolecular closure problem", recovering unresolved interactions beyond classical force fields while preserving thermodynamic consistency and mechanistic interpretability. We examine theoretical foundations, tools and frameworks, computational trade-offs, and unresolved issues, including model expressiveness and stability. We outline prospective research avenues at the intersection of machine learning, statistical physics, and computational chemistry, contending that future advancements will depend on mechanistic inductive biases, and integrated differentiable physical learning frameworks for biomolecular simulation and discovery.
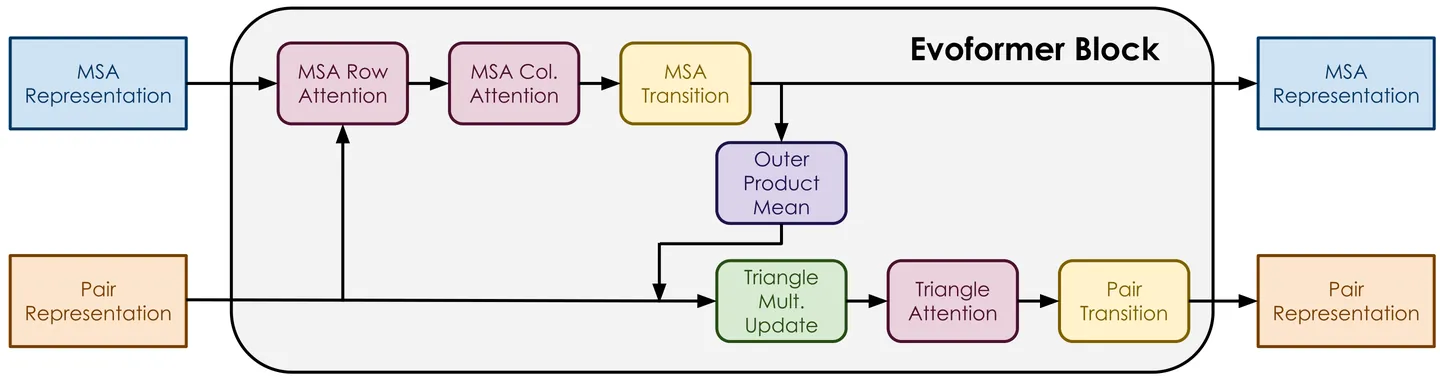
Models such as AlphaFold2 and OpenFold have transformed protein structure prediction, yet their inner workings remain poorly understood. We present a methodology to systematically evaluate the contribution of individual OpenFold components to structure prediction accuracy. We identify several components that are critical for most proteins, while others vary in importance across proteins. We further show that the contribution of several components is correlated with protein length. These findings provide insight into how OpenFold achieves accurate predictions and highlight directions for interpreting protein prediction networks more broadly.
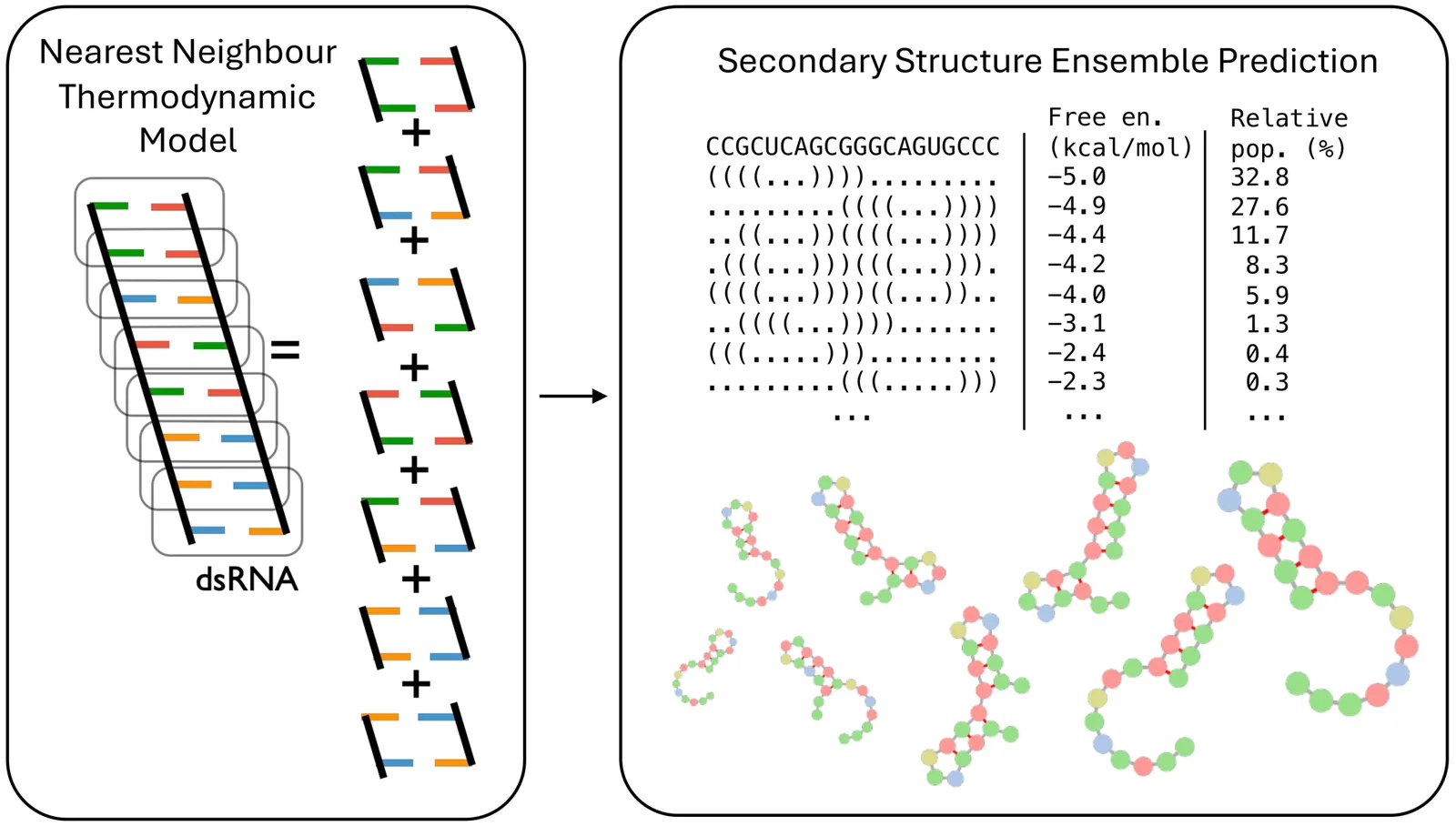
Predicting the secondary structure of RNA is a core challenge in computational biology, essential for understanding molecular function and designing novel therapeutics. The field has evolved from foundational but accuracy-limited thermodynamic approaches to a new data-driven paradigm dominated by machine learning and deep learning. These models learn folding patterns directly from data, leading to significant performance gains. This review surveys the modern landscape of these methods, covering single-sequence, evolutionary-based, and hybrid models that blend machine learning with biophysics. A central theme is the field's "generalization crisis," where powerful models were found to fail on new RNA families, prompting a community-wide shift to stricter, homology-aware benchmarking. In response to the underlying challenge of data scarcity, RNA foundation models have emerged, learning from massive, unlabeled sequence corpora to improve generalization. Finally, we look ahead to the next set of major hurdles-including the accurate prediction of complex motifs like pseudoknots, scaling to kilobase-length transcripts, incorporating the chemical diversity of modified nucleotides, and shifting the prediction target from static structures to the dynamic ensembles that better capture biological function. We also highlight the need for a standardized, prospective benchmarking system to ensure unbiased validation and accelerate progress.